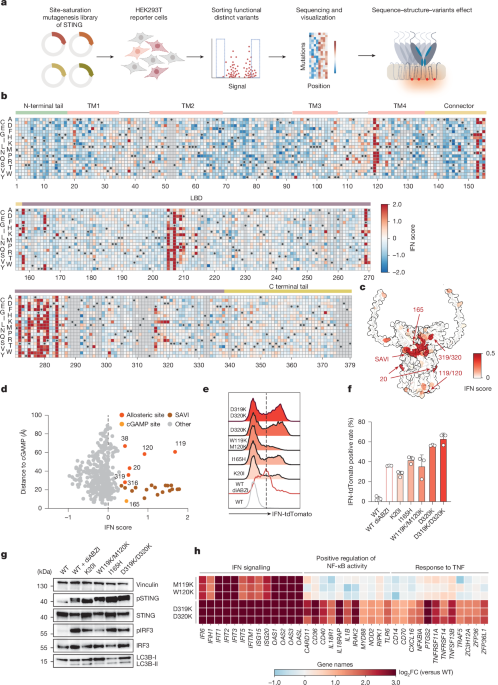
MainIn mammalian cells, STING is expressed as a dimeric transmembrane (TM) protein at the endoplasmic reticulum and orchestrates innate immune responses to 2′3′-cGAMP, the endogenous product of the DNA sensor cGAS, bacterial cyclic dinucleotides and immunotherapeutic agents18,19,20,21,22. Ligand engagement of the cytosolic ligand-binding domain (LBD) triggers a profound conformational rearrangement that drives STING oligomerization, enabling dedicated protein–protein interactions essential for the initiation of immune signalling4,5,6,8,23. In addition to these structural transitions, cellular activation is also tightly linked to intracellular trafficking: activated STING translocates from the endoplasmic reticulum to the Golgi and post-Golgi compartments, where heteromeric signalling complexes assemble8,9,10,24,25. STING activation culminates in the upregulation of type I interferon (IFN) and NF-κB responses and the concomitant triggering of a non-canonical form of autophagy characterized by conjugation of LC3B to single membranes26,27,28 (Extended Data Fig. 1a). Whereas transcriptional responses are governed by the recruitment of TBK1, LC3B lipidation was recently shown to depend on STING serving as a proton channel in post-Golgi membranes12,13.High-resolution structures of STING capturing both inactive and active states, revealed conformational transitions in the LBD and connector region, as well as extensive remodelling at the dimer–dimer interface on ligand binding4,23,29. Besides ligand-induced activation, STING trafficking and signalling are also prompted by missense mutations. In the autoinflammatory syndrome STING-associated vasculopathy with onset in infancy (SAVI), autoactive gain-of-function (GOF) mutations trigger profound inflammatory reactions plausibly by mimicking or stabilizing active-state conformations30,31,32,33,34,35,36,37. In addition, single-site substitutions can also enhance STING activation through, for example, suppressing inhibitory protein–protein interactions22,29,38. However, in aggregate, annotated STING variants at present comprise only a minor fraction of the full mutational spectrum of STING. We proposed that mapping the entire possible sequence space of STING onto cellular phenotypes would uncover previously unrecognized principles of STING biology.Discovery of autoactive STING variantsWe combined a deep mutational scanning (DMS) approach covering every amino-acid substitution with an IFN reporter assay in human embryonic kidney 293T (HEK293T) cells39,40,41 (Fig. 1a). To establish a screening workflow, we tested a previously characterized SAVI-associated GOF allele (STING(N154S))32. Inducible expression of the SAVI STING variant triggered robust IFN reporter activity, whereas no discernible response was observed for wild-type STING in the absence of a ligand or for a STING variant defective in IRF3 activation (S366A) (Extended Data Fig. 1b). The level of reporter activity induced by the SAVI variant was equivalent to stimulation of ectopically expressed wild-type STING with the chemical agonist diABZI20 (Extended Data Fig. 1b). Thus, these findings validate the dynamic range of our reporter system and establish an experimental framework for unbiased discovery of functional STING variants.Fig. 1: A site-saturation mutagenesis screen defines single amino-acid substitutions enabling STING-induced type I IFN.a, Workflow to discover STING variants with altered function. b, Heatmap of variant effects on IFN induction. Colour scale denotes IFN induction scores from −2 (blue; decreased relative to WT) to 0 (white; WT) to 2 (red; increased relative to WT). Wild-type (WT) amino acids and missing data (Methods) shown as black dots and grey boxes, respectively (representative of n = 2). c, AlphaFold model of the human STING (hSTING) homodimer coloured by the average IFN score at each site to reflect mutation tolerance. Colour scale denotes IFN induction scores ranging from 0 (white; WT) to 0.5 (red; increased relative to WT). d, Per-position average IFN score plotted against distance to bound cGAMP, with cGAMP ligand-binding sites, allosteric sites and SAVI cluster highlighted. e, IFN response from HEK293T IFN reporter cells. Cells were seeded at 0.2 million per condition and transfected with indicated enhanced green fluorescent protein (eGFP)-tagged STING variants for 24 h. At least 10,000 events were acquired per sample for flow cytometry analysis. f, Quantification of IFN reporter activity corresponding to the conditions shown in e (n = 3 biological replicates). Data are mean ± s.d. from three biological replicates. g, Immunoblot analysis of pSTING, STING, pIRF3, IRF3 and LC3B-I/II in IFN reporter cells seeded at 0.2 million per well and expressing indicated variants for 24 h, followed by dimethylsulfoxide (DMSO) or diABZI treatment. Vinculin is used as loading control (representative of n = 3). h, Differentially expressed genes identified from bulk RNA-seq of STING1 knockout THP-1 cells reconstituted with inducible eGFP-tagged wild-type STING, STING(W119K/M120K) or STING(D319K/D320K), seeded at 1 million cells per sample and induced with doxycyline for 24 h before RNA collection. The heatmap shows IFN-stimulated genes (ISGs) and broader inflammatory markers from the total list of differentially expressed genes (false discovery rate (FDR) < 0.05, |log2[FC]| > 1, n = 3 biological replicates, details in Extended Data Fig. 3a). FC, fold change. Panel a created in BioRender; Zhang, B. https://biorender.com/jv5hnal (2025).We first conducted a saturation mutagenesis screen with the goal to define autoactive variants of STING driving IFN reporter expression in the absence of a ligand. Sequencing of the input population recovered 7,189 out of 7,202 (99.82%) possible genotypes, confirming good representation of each variant (Extended Data Fig. 1c). To minimize bias, we filtered out variants with insufficient reads and eventually acquired 6,251 out of 7,202 (86.80%) genotypes. Subsequent analysis and heatmap representation revealed a broad continuum of activities, ranging from silent substitutions to strongly autoactive alleles with IFN scores well correlated among independent replicates (Fig. 1b and Extended Data Fig. 1d,e). Notably, the discovered GOF variants were not randomly distributed, but instead clustered within discrete structural regions, including the connector loop (bridging the TM domain with the LBD), the tetramer interface and the LBD (Fig. 1b,c). Some hotspots within these clusters not only overlapped with but also extended beyond mutations previously reported in individuals with SAVI, thereby validating the approach and expanding the known mutational and functional diversity for these structural regions30,31,32,33,34,35,37. Several constitutively active variants mapped to previously unrecognized allosteric sites spatially separated from the cGAMP-binding site (Fig. 1d). These included residues in the TM1 (K20), in the luminal loop connecting TM3 and TM4 (W119/M120), and within an unstructured C-terminal loop of the LBD (E316/D319/D320). In addition, substitutions of I165 within the cGAMP-binding cleft, also mark active variants. Individual testing confirmed robust reporter activity on expression of these STING variants, mirroring the graded activities recorded in the screen and without altering overall protein abundance (Fig. 1e–g and Extended Data Fig. 1f–h). Apart from these main activating clusters, a set of scattered variants, including Y46R, R169K and P371G, also confirmed IFN-inducing capacity (Extended Data Fig. 1g). Together, these results reveal a widely distributed phenotypic landscape of STING, demonstrating that diverse structural regions contribute to finely graded control of its IFN signalling potential.In addition to transcriptional induction of IFN, STING also engages the more ancient non-canonical autophagy pathway7,9,27. To determine whether the autoactive property across these two effector branches is coordinated, we next coupled saturation mutagenesis of STING to quantitative assessment of LC3B lipidation42 (Extended Data Fig. 2a–d). Notably, IFN and LC3B lipidation scores were correlated: STING variants with heightened IFN responses, accompanied by signalling hallmarks linked to IFN induction, such as phosphorylation of STING and IRF3, also showed LC3B lipidation responses (Fig. 1g and Extended Data Fig. 2e–g). However, despite similar constitutive activity, the relative balance of IFN versus LC3B lipidation activity diverged for certain variants (for example, W119K/M120K), indicating that STING mutations can skew the partitioning of signalling output.To monitor the full spectrum of transcriptional activities induced by constitutively active STING proteins in immune cell-relevant contexts, we reconstituted STING1-deficient THP-1 monocytes with wild-type, W119K/M120K or D319K/D320K variants and performed bulk RNA sequencing (RNA-seq). Unbiased transcriptomic analysis showed strong enrichment of IFN-associated gene expression programs (‘IFN alpha/beta signalling’) across both variants (Fig. 1h and Extended Data Fig. 3a–c). However, only the D319K/D320K mutant, but not the W119K/M120K variant, also showed enrichment for inflammatory gene categories linked to NF-κB signalling (Fig. 1h and Extended Data Fig. 3a–c). We confirmed that both variants induced activation of IFNB1 and the IFN-stimulated genes IFIT1 and IFIT3, whereas only D319K/D320K upregulated NF-κB targets such as CCL3 and CCL4, along with enhanced IL-8 production (Extended Data Fig. 3d–g). Together, these results delineate the diverse signalling landscape of autoactive STING variants, revealing new autoinhibitory residues and demonstrating that discrete substitutions within the protein can qualitatively rewire STING signalling outputs.Architecture of autoactive STING variantsTo explain how STING can drive constitutive downstream signalling, we determined cryogenic-electron microscopy (cryo-EM) structures of variants harbouring spatially divergent substitutions (Extended Data Figs. 4 and 5a–c). One cluster of autoactivating mutations, centred at residues E38, W119, M120 and Y106, caught our attention as it highlights the luminal part of STING facing the endoplasmic reticulum interior as a potent regulatory element. A 2.9-Å resolution cryo-EM reconstruction of the W119K/M120K mutant revealed elongated, curved filamentous oligomers with clear lipid densities covering the TM domains (Fig. 2a,b). The W119K/M120K bent filaments each comprise around 30–60 parallel stacked STING dimers that assemble into crescent-like structures with more than 65 nm diameter (Fig. 2b). Consistently, on cellular expression of the W119K/M120K variant, we observed extended filamentous assemblies (Extended Data Fig. 5d). Structural modelling revealed that the two lysine substitutions neutralize the repulsive force at the luminal interface and form new electrostatic contacts with E38 and Y106 in the dimer unit, drawing the luminal ends of TM3 and TM4 towards TM1 of the opposing protomer and displacing the cytosolic end of TM3 outwards (Fig. 2c,d). This rearrangement propagates to rotate the LBD, resulting in a conformation in which the STING dimer features an untwisted linker orientation between the TM and cytosolic domains closely resembling cGAMP-bound STING structure29 (Protein Data Bank (PDB) code 8IK3, global root mean-square deviation of 1.3 Å). Consistent with this mechanism, the substitutions E38K and Y106K markedly reduced the ability of STING(W119K/M120K) to trigger IFN signalling (Extended Data Fig. 5e,f). These data thus support a model in which the luminal residues W119 and M120 maintain STING autoinhibition through preventing structural compression of the TM domains to restrain the formation of STING protein filaments.Fig. 2: Structural mechanism of select autoactive STING substitutions.a, Cryo-EM density map of a STING(W119K/M120K) filament. b, Representative micrograph image for STING(W119K/M120K) variant from 16,001 micrographs (left) and close-up view of the higher-order polymer assembled from the aligned decameric EM density (right). c, An isolated STING(W119K/M120K) dimer (left) and close-up view comprising the dimer interface (right), with red arrows indicating the helical displacement relative to the wild-type conformation. d, Radar plots showing the IFN scores of all mutations for the E38, Y106, W119 and M120 positions. Higher radial values indicate stronger intrinsic self-activation. e, Radar plots showing the IFN scores of all mutations for the P317, A318, D319 and D320 positions. Higher radial values indicate stronger intrinsic self-activation. f, Cryo-EM density map of the STING(D319K/D320K) filament complex. g, Representative micrograph images for STING(D319K/D320K) variant from 21,613 micrographs (left) and close-up view of the higher-order polymer assembled from the aligned octameric EM density (right). White arrows highlight the oligomeric STING particles. h, Ribbon diagram of STING(D319K/D320K) tetramer. i, AlphaFold3 prediction of a wild-type STING tetramer with interface shown as a semitransparent surface coloured by electrostatic potential and overlaid with a ribbon diagram. j, Cryo-EM density map of the STING(I165H) dimer. k, Isolated wild-type STING (left) and STING(I165H) dimer (right), and close-up view of the dimer interface (far right). Red arrows highlight the conformational changes. l, Radar plots showing the IFN scores of all mutations for the I165 position. Higher radial values indicate stronger intrinsic self-activation. Scale bars, 20 nm (b,g).Another functionally relevant hotspot maps to a flexible loop spanning residues 317–320 that shows striking basic sensitivity, as only lysine or arginine substitutions activated STING (Fig. 2e). The cryo-EM structure of the D319K/D320K variant revealed a planar oligomeric assembly in which an unaltered, apo-like TM bundle is paired with a rotated active-like LBD yielding a conformation distinct from previously reported STING structures (Fig. 2f and Extended Data Figs. 4g and 5g,h). Although K319 and K320 are not well resolved in our cryo-EM maps, AlphaFold3 modelling of oligomeric apo wild-type STING places the D319/D320 loop in proximity to an acidic helix of the neighbouring dimer, creating a potentially repulsive contact that is relieved in the active state by the curved geometry of the cGAMP-bound assembly5,29 (Fig. 2h,i and Extended Data Fig. 4n). Consistent with an electrostatic mechanism, charge-reversing mutations to residues E339 and E340 at the opposite helix abolished constitutive activity of STING(D319K/D320K) in cells (Extended Data Fig. 5i,j). How charge reversal at this interface is transmitted to LBD rotation remains to be defined.Residue 165 represents another notable mutation-sensitive site that is uniquely positioned close to the cGAMP-binding pocket. Cryo-EM analysis of the I165H mutant reveals a dimer resembling apo STING, with no major TM or LBD rearrangements, but localized structural changes (Fig. 2j,k). Substituting I165 with acidic or aromatic residues introduces favourable interactions with R169 and Y274, stabilizing a more closed LBD conformation while releasing the twist of the connector helix towards the TM region (Fig. 2k,l). This H165–R169 interaction further remodels the helix near Y274 into a cGAMP-activated loop conformation, plausibly contributing to STING hyperactivation. Consistently, introducing R169A into the I165H background abolished IFN signalling (Extended Data Fig. 5k,l). Similar to previous ligand-bound and SAVI-mutant STING structures36, the I165H substitution favours a pre-activated dimer conformation whose progression to a fully active oligomeric state probably requires a native membrane environment.We further identified an activation-sensitive element within the TM hetero-oligomeric interface, at which mutations that attract a closely positioned residue pair, K20 and L93, led to a pronounced autoactive signature in our screen and conferred constitutive signalling on validation (Extended Data Fig. 5m–o). In wild-type STING, these residues have co-evolved to remain spatially separated, thereby preventing aberrant contact (Extended Data Fig. 5p).We next combined cellular reconstitution with downstream signalling analysis to examine how mutation-induced bespoke structural arrangements confer STING-mediated cellular activation. Confocal microscopy revealed that both STING variants I165H and D319K/D320K, which comprise LBD mutations, showed phosphorylated STING (pSTING) puncta colocalizing with trans-Golgi network (p230) and endolysosomal (LAMP1) markers, indicative of constitutive endoplasmic reticulum-to-Golgi trafficking (Extended Data Fig. 6a,b). Disruption of the Golgi with Brefeldin A abolished the intrinsic activity of these variants, reflecting their dependence on Golgi translocation for signalling initiation, similar to natural STING activation (Extended Data Fig. 6c–f). By contrast, pSTING(W119K/M120K) showed only limited overlap with the trans-Golgi network marker p230, suggesting that its ability to form extended curved filaments enables IFN signalling independently of the Golgi membrane context. Consistently, activation of signalling downstream of STING(W119K/M120K) remained largely unaffected by brefeldin A (Extended Data Fig. 6g,h). Together, these results show that STING is held inactive by a distributed set of structural constraints, the release of which drives distinct signalling-competent states that dictate cellular responses (Extended Data Fig. 6i).Inhibitory STING variants identified at scaleTo systematically define sequence determinants of STING activity in the presence of a ligand, we extended our saturation mutagenesis approach to map variants that modulate cGAMP-initiated responses (Extended Data Fig. 7a). Comparing STING mutants in the IFN reporter assay with or without cGAMP stimulation revealed a constitutively hyperactive cluster near known SAVI sites, as well as a distinct set of cGAMP-dependent substitutions unexpectedly positioned within endoplasmic reticulum-luminal linkers and pockets that accommodate the agonist C53 (ref. 23) (Fig. 3a,b). We validated that mutations at the luminal end of STING selectively amplified IFN signalling in the presence of cGAMP, suggesting that the LBD structurally communicates to this region to allosterically tune ligand-driven activation (Extended Data Fig. 7b).Fig. 3: Activating and inhibitory STING variants in the presence of cGAMP.a, Scatter plot comparing IFN scores from STING mutagenesis screens with or without cGAMP stimulation. b, Wild-type STING structure determined in-house (PDB 9SWM), coloured by the number of mutations at each site showing cGAMP-dependent GOF activity. c, AlphaFold structure of the hSTING homodimer coloured by residue-wise IFN scores on cGAMP stimulation, reflecting mutation tolerance (left). Colour scale denotes IFN scores ranging from −1 (blue; decreased relative to WT) to 0 (white; WT). A close-up view of residue S80 is shown, coloured by the mutation counts showing discordance between the experimental IFN scores and the AlphaMissense pathogenicity prediction. d, Correlation between the IFN scores and the AlphaMissense pathogenicity scores. The red box marks regions of discordance between the two scores, and the blue box highlights regions of concordance. e, Histogram depicting selected sites classified as ‘match’ or ‘mismatch’ on the basis of concordance between their IFN scores and AlphaMissense pathogenicity scores. f, IFN reporter activity of the STING S80 variants in IFN reporter cells seeded at 0.2 million per well and treated with diABZI (n = 3 biological replicates). Data are mean ± s.d. from three biological replicates. P values were obtained using one-way ANOVA followed by Tukey’s multiple-comparisons test and ordinary two-way ANOVA. g, Immunoblot analysis of pSTING, STING, pIRF3, IRF3, pTBK1, TBK1 and LC3B-I/II in IFN reporter cells seeded at 0.2 million per well, transfected with plasmid encoding wild-type STING or S80 variants and treated with DMSO or diABZI (representative of n = 3). h, WebLogo plot showing the evolutionary conservation at each position, with letter height proportional to the amino acid frequency.Conversely, loss-of-function (LOF) variants under cGAMP stimulation mapped to canonical regulatory features, including the cGAMP-binding pocket, the lid region covering cGAMP and the C-terminal tail motif required for TBK1 and IRF3 recruitment5,6,8 (Fig. 3c and Extended Data Fig. 7c). Beyond these expected regions, two extra mutationally intolerant clusters emerged at residues I10, R14, E149 and within a second region spanning residues 78–80 (Fig. 3c). Because reduced protein abundance is a common mechanism underlying pathogenic missense variants, we asked whether the identified LOF effects reflected structural destabilization rather than signalling-specific defects. To disentangle these possibilities, we compared our experimental IFN activity scores with predictions from three independent computational variant effect predictors43,44,45. Positions such as I10 and E149 showed good agreement between predicted pathogenicity and experimentally observed loss of activity, possibly due to a structural role in stabilizing the LBD architecture (Fig. 3d,e and Extended Data Fig. 7d–f). By contrast, we uncovered a second class of LOF variants in which experimental data and predictions diverged (Fig. 3d). These substitutions were computationally predicted to be neutral, yet impaired signalling in cells. These discordant positions were strongly enriched in surface-exposed, unstructured loops, regions where flexibility probably limits the predictive performance of current structural models (Fig. 3c–e and Extended Data Fig. 7g,h). For instance, residue S80 in the TM2–TM3 loop showed pronounced sensitivity to hydrophobic substitutions and orthologue analysis across 184 vertebrate STING sequences revealed negative selection against hydrophobic residues at this site, underscoring the importance of loop flexibility for activation (Fig. 3f–h). More broadly, positions with low experimental IFN activity tended to coincide with evolutionarily conserved residues, supporting the view that mutationally constrained elements encode critical regulatory functions (Extended Data Fig. 7i). Together, these results define a comprehensive LOF map of STING activation, with canonical structural determinants aligning well with computational predictions, whereas flexible loop regions that modulate signalling competence are systematically underpredicted.Uncoupling STING-dependent IFN and autophagy responsesTo systematically explore the relationship between IFN and LC3B lipidation responses, and to distinguish transcriptional from non-transcriptional regulatory elements, we performed an extra screen for ligand-induced LC3B lipidation (Extended Data Fig. 8). Integrating the obtained LC3B scores with corresponding IFN values enabled the construction of a combined sequence-function map that resolved distinct categories of signalling behaviour (Fig. 4a). STING variants showed a wide range of activities, including divergent phenotypes in which mutations selectively enhanced or abolished IFN or LC3B responses. To categorize residues according to their dominant regulatory roles, we vectorized the counts of mutations for each residue distributed across the corners in the sequence-function map (Fig. 4b). These residue-level data were projected into a two-dimensional phenotypic space, positioning residues according to their combined IFN and LC3B activities (Fig. 4c). This analysis revealed discrete functional clusters composed of residues that govern distinct aspects of ligand-induced STING activation and regulation (Fig. 4c,d). When mapped onto the three-dimensional STING structure, these clusters grouped into spatially organized regions that preferentially modulate transcriptional (that is, IFN) versus non-transcriptional (that is, LC3B lipidation) outputs (Fig. 4d,e and Extended Data Fig. 9a–c). For example, the C-terminal tail is a hotspot for IFN-defective variants, consistent with its role in TBK1 and IRF3 recruitment and its dispensability for LC3B lipidation5,7. We also identified discrete structural regions composed of variants that selectively compromised LC3B lipidation while preserving IFN activation and these were localized to TM3, the LBD tip and an adjacent C-terminal helix. We validated that substitutions within the LBD tip selectively impaired LC3B lipidation (Extended Data Fig. 9d,e). Moreover, a single mutation in TM3 (L100E) nearly abolished STING-induced LC3B conjugation, yet left IFN activation intact after stimulation with diABZI (Fig. 4f–i). Confocal microscopy revealed that L100E STING forms ligand-induced foci that fail to traffic beyond the ER, thereby preventing the membrane remodelling and proton flux required for LC3B lipidation (Extended Data Fig. 9f). Under physiological conditions, STING trafficking is essential for both LC3B lipidation and efficient IFN induction. However, L100E stabilizes STING oligomers within the ER, enabling IFN production in the absence of trafficking. This finding demonstrates that the oligomerization capacity of STING, and not the subcellular localization per se, dictates IFN signalling, whereas LC3B lipidation strictly requires endoplasmic reticulum exit and subsequent membrane remodelling. The trafficking-blocking property of L100E also acted in trans: co-introduction of L100E suppressed the IFN-inducing capacity of the trafficking-dependent D319K/D320K variant, but had no effect on the trafficking-independent W119K/M120K mutant (Fig. 4i and Extended Data Fig. 9g–j). We next leveraged the trafficking defect of the L100E mutant to examine the spatial requirements of NF-κB activation, another transcriptional output of STING signalling. Stimulation of L100E in NF-κB-sensitive THP-1 cells failed to elicit a detectable NF-κB response, as measured by CCL3 expression, despite robust induction of type I IFN signalling (Extended Data Fig. 10c). This observation supports a model, in which the NF-κB transcriptional program requires post-endoplasmic reticulum trafficking of STING46. In line with this interpretation, the endoplasmic reticulum-active W119K/M120K mutant, which strongly induces IFN, showed only minimal NF-κB activity (Extended Data Fig. 10d). Collectively, these results provide a systematic functional map linking individual STING residues to discrete transcriptional and non-transcriptional outcomes. They identify molecular determinants that preserve IFN competence while selectively abolishing LC3B lipidation, an uncoupling not previously achieved by mutation, and reveal that STING trafficking is a shared prerequisite for both LC3B lipidation and NF-κB activation under normal conditions, yet dispensable for IFN induction in the context of endoplasmic reticulum-confined STING oligomerization.Fig. 4: Molecular determinants of cGAMP-mediated STING responses.a, Correlation between IFN and LC3B scores. Red, IFN + LC3B percentile greater than 170%; blue, IFN + LC3B percentile less than 30%; green, IFN greater than 70% and (IFN-LC3B) greater than 34%; yellow, LC3B greater than 70% and (LC3B-IFN) greater than 34%. b, Workflow for uniform manifold approximation and projection-based projection. c, Uniform manifold approximation and projection (UMAP) of STING variants. d, Histogram depicting response preferences across the entire protein, with peak heights corresponding to the number of variants at each site. e, AlphaFold structure of the hSTING homodimer coloured by the mutation counts per site from d with a threshold from 3 to 10. f,g, IFN reporter activity (f) and eGFP-LC3B reporter activity (g) of wild-type STING and L100E variants in reporter cells seeded at 0.2 million per well following diABZI stimulation. At least 10,000 cells were acquired per sample for flow cytometry analysis. h, Immunoblot analysis of pIRF3, STING and LC3B-I/II in IFN reporter cells were seeded at 0.2 million per well and expressing wild-type STING or L100E variants for 24 h, followed by DMSO or diABZI treatment (representative of n = 3). i, HEK293T stably expressing a ratiometric super-ecliptic pHluorin (SEP) and mRuby3 reporter localized to GALT transfected with selected STING variants and stimulated or not with diABZI. Violin plot showing the ratio of SEP/mRuby3 calculated from more than 8,000 cells (representative of n = 3). Data are mean ± s.d. from three biological replicates (f,g). P values were obtained using one-way ANOVA followed by Tukey’s multiple-comparison test and ordinary two-way ANOVA (f,g,i).Intersection of structural states with functional residuesLigand-induced STING activation is accompanied by profound structural rearrangements that couple ligand binding to signalling4. To relate these transitions to our functional dataset, we integrated DMS measurements with high-resolution apo (PDB 9SWM) and cGAMP-bound (PDB 8IK3)29 STING structures. The magnitude of residue displacement (that is, α-carbon atom movement) between states showed no correlation with functional relevance and extensive contact change alone was likewise insufficient to explain functional effects, as variants showing large contact differences frequently remained functionally neutral (Extended Data Fig. 10a). Instead, functional residues (average GOF > 0.25) combine structural displacement with interaction rewiring. Mapping these features onto the STING structure revealed that such residues cluster at two opposing poles of STING: the luminal region and the LBD (Extended Data Fig. 10b). By contrast, residues undergoing substantial contact rearrangements but remaining functionally neutral localize predominantly to intervening regions such as the connector region and TM3 (Extended Data Fig. 10b). This spatial organization indicates that cGAMP-induced STING activation is mediated by mechanically coupled functional sites positioned at both poles of the protein, which are linked by an intervening network of residues that primarily acts as a structural scaffold transmitting conformational changes across the molecule.Implications on clinical aspectsSTING1 mutations found in individuals with SAVI show notable variability in clinical presentation and incomplete penetrance, complicating their clinical interpretation31,32,47,48. Beyond the well-characterized benign or pathogenic alleles (benign or benign-like and pathogenic or pathogenic-like), large-scale sequencing has uncovered various variants of uncertain significance within the human population49,50. To systematically assess their functional impact, we integrated our variant screen with the ClinVar reference dataset. Functional IFN scores accurately segregated known variants according to their benign or pathogenic classification, validating the predictive capacity of our assay (Fig. 5a). Among variants of uncertain significance, our dataset identified substitutions within established SAVI hotspots, as well as W119C, a constitutively active STING variant mapping outside all previously defined SAVI sites (Fig. 5a). Similarly, cross-referencing gnomAD entries with our maps nominated GOF variants, including establised SAVI hotspots as well as W119C and P317R (Fig. 5b). Expression of either mutant was sufficient to trigger ligand-independent type I IFN signalling, confirming their hyperactive nature (Fig. 5c). While examining more STING1 variants of uncertain significance, we identified an individual with a SAVI-like clinical presentation, including features of interstitial lung disease and persistently enhanced type I IFN signalling (as measured by the expression of a panel of interferon-stimulated genes (ISGs) in whole blood)51 (Fig. 5d, Extended Data Fig. 10e and Methods). Next-generation panel sequencing identified a heterozygous STING1 variant c.956A>G (p.(Asp319Gly)) (Extended Data Fig. 10f and Methods). This substitution is extremely rare, being absent from any of 1,612,180 alleles annotated on gnomAD. Analogous to the D319K mutant characterized above, substitution of aspartic acid with glycine at position 319 is predicted to neutralize the charge of the flexible loop. Multiple-sequence alignment of 184 STING1 sequences revealed that the flexible loop at position 319 is highly conserved, with net acidity preserved across species (Extended Data Fig. 10g,h). To test the functional impact of charge neutralization of a substitution of D319, we reconstituted D319G in THP-1 STING1 knockout cells and observed activation of all three branches of STING signalling, including transcriptional induction of type I IFN and NF-κB-dependent genes as well as LC3B lipidation (Fig. 5e,f and Extended Data Fig. 10i). Together, these findings identify residue 319 as a previously unrecognized regulatory site connecting a rare human genetic variant to STING hyperactivation.Fig. 5: STING saturation mutagenesis enable variant interpretation.a, Violin plot depicting the average IFN scores of STING variants categorized in ClinVar database. B/BL, benign or benign like; P/PL, pathogenic or pathogenic like; VUS, variant of uncertain significance. b, Scatter plot depicting the average IFN and LC3 scores of STING variants characterized in the gnomAD database. c, IFN reporter activity of indicated Flag-tagged STING variants in IFN reporter cells seeded at 0.2 million per well before transfection. At least 10,000 cells were acquired per sample for flow cytometry analysis. d, IFN score of an index patient with heterozygous STING1 c.956A>G (p.Asp319Gly) variant. e, mRNA expression levels of IFNB1, IFIT3 (ISGs) and CCL3, CCL4 (NF-κB-dependent) in STING1 knockout THP-1 cells reconstituted with inducible wild-type STING or D319G variant and seeded at 0.5 million per well, following DMSO or diABZI stimulation. f, Immunoblot analysis of pIRF3 and LC3B-I/II in IFN reporter cells transfected with plasmid encoding wild-type STING or D319G variant and treated with DMSO or diABZI. g, Scatter plot of STING mutations from the COSMIC database depicting mutation counts and IFN scores with cGAMP stimulation. The mutations with count of three or more and IFN score less than zero are highlighted in cyan. h, Kernel density estimation of COSMIC mutations (count three or more, cyan) and the entire mutational library (grey) corresponding to IFN scores with cGAMP. P values were calculated using the Mann–Whitney U-test. i, IFN reporter activity of indicated eGFP-tagged STING variants transfected into IFN reporter cells seeded at 0.2 million per well, followed by DMSO or diABZI treatment. Data are mean ± s.e.m. from three biological replicates (c,e,i). P values were obtained using one-way ANOVA followed by Tukey’s multiple-comparison test and ordinary two-way ANOVA (c,e,i).We next focused on the clinical implications of the LOF DMS dataset and analysed variants reported in the COSMIC database, representing somatic mutations observed in human cancers52. Cancer-associated variants showed a global shift towards reduced IFN activity, with an enrichment of LOF alleles within the cGAMP-binding pocket (G230A, R293Q, R232Y and R169W) and within our newly identified mutationally intolerant cluster (R14W) (Fig. 5g,h). This pattern suggests that attenuation of cGAMP sensing, potentially downstream of DNA-triggered immune surveillance, may confer a selective advantage during tumour evolution. Indeed, representative alleles rendered cells unresponsive to STING agonists, confirming their functional silencing (Fig. 5i). Together, these results demonstrate that DMS-based functional immune mapping provides a powerful framework to expedite the interpretation of clinical variants and reveal how human STING diversity shapes immune signalling across autoinflammatory and malignant contexts.ConclusionOur study defines the sequence–function landscape of STING, providing a comprehensive framework for understanding how single amino-acid changes remodel the conformational and signalling properties of this central innate immune adaptor. Extending beyond previously characterized disease-associated alleles and a restricted set of IFN-affecting substitutions in mouse STING22, our forward mutational screen reveals that the STING protein architecture is finely balanced at the edge of higher-order oligomerization: a design principle that both enables rapid immune activation and predisposes to spontaneous signalling when perturbed. Structural analysis of hyperactive variants further demonstrates that distinct filament geometries correlate with different signalling states. Whereas ligand-bound STING adopts moderately curved filaments, the autoactive W119K/M120K variant forms highly curved assemblies that closely resemble the activated cGAMP-bound conformation and can initiate signalling directly from the endoplasmic reticulum membrane. By contrast, the trafficking-dependent D319K/D320K mutant assembles into comparatively straighter oligomers that seem to represent a primed but not fully active state. These assemblies probably require the distinct lipid composition and membrane curvature encountered at post-endoplasmic reticulum compartments, such as the Golgi or endolysosomal membranes, to transition into fully signalling-competent structures. Together, these findings suggest that productive STING signalling is governed not only by oligomerization per se but also by the precise geometry of filament assembly and its compatibility with the surrounding membrane environment53,54,55,56.In parallel, our mutational data provide a mechanistic model for understanding how STING coordinates distinct downstream responses from different subcellular compartments. The separation of type I IFN signalling, LC3B lipidation and NF-κB activation is determined by two intersecting parameters: the oligomeric state of the STING filament and its subcellular localization. Type I IFN induction is governed primarily by the formation of high-curvature STING filaments and does not strictly require Golgi translocation, as exemplified by the W119K/M120K variant, which potently triggers IFN signalling from the endoplasmic reticulum while showing only limited anterograde trafficking. LC3B conjugation, by contrast, depends on STING reaching post-endoplasmic reticulum compartments, yet this requirement can be satisfied by even a minor trafficked protein fraction, consistent with Brefeldin A abolishing LC3B lipidation but not IFN induction downstream of W119K/M120K. NF-κB activation imposes the most stringent demands: it requires both Golgi translocation and sufficient accumulation of STING at the trans-Golgi network to surpass a higher activation threshold. This is directly supported by the trafficking-deficient L100E mutant, which robustly activates type I IFN but fails to induce NF-κB on ligand stimulation. Together, these findings redefine intracellular trafficking not merely as a route for STING activation, but as a process capable of differentially gating its transcriptional and non-transcriptional outputs according to both filament geometry and the extent of subcellular redistribution.Beyond elucidating mechanistic principles, our comprehensive variant map rationalizes naturally occurring polymorphisms and clinically reported variants of uncertain significance, providing a resource to interpret their functional impact. From a synthetic-biology perspective, the engineered hyperactive variants described here offer an opportunity to program cell states with tuneable innate immune properties and to design immune sensors with defined effector bias57. Our work establishes a scalable strategy to decode the molecular principles of immune signalling proteins and illustrates how deep mutational interrogation can reveal the intrinsic rules that govern activation and regulation of host defence systems. More broadly, our approach creates a high-resolution map that links mutational perturbations with cellular phenotypes, providing new dimensions to our understanding of protein function that could be used for the study of many other proteins in their cellular context58,59,60.MethodsCell cultureHeLa (CCL-2) cells were obtained from Sigma-Aldrich. HEK293T cells were a gift from D. Trono, originally purchased from the American Type Culture Collection. BJ-5ta cells were obtained from the American Type Culture Collection. THP-1-Dual STING1 knockout cells were purchased from Invivogen (thpd-kostg). HeLa and HEK293T cells were cultured in Dulbecco’s Modified Eagle medium (DMEM) (Thermo Fisher Scientific, 41965039) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS) (Thermo Fisher Scientific, Gibco SKU, 10270106), 1% (v/v) penicillin (100 IU ml−1)–streptomycin (100 μg ml−1) (BioConcept, 4-01F00-H), 2 mM l-glutamine (Thermo Fisher Scientific, 25030024). BJ-5ta cells were cultured in a mixed medium consisting of 80% of DMEM and 20% of Medium 199 (Thermo Fisher Scientific, 41150087) supplemented with 10% FBS, 2 mM l-glutamine and 1% (v/v) penicillin–streptomycin. THP-1 cells were cultured in Roswell Park Memorial Institute 1640 Medium (RPMI 1640) (Thermo Fisher Scientific, 21875034) containing 10% (v/v) heat-inactivated FBS, 25 mM HEPES (BioConcept, 5-31F00-H), 2 mM l-glutamine and 1% (v/v) penicillin–streptomycin. All cells were cultured at 37 °C and at atmospheric O2 and 5% CO2. HeLa cGAS knockout cells were reported previously38,61. Cell lines were routinely tested and confirmed to be negative for mycoplasma contamination.PlasmidsThe pEFBos-eGFP-STING and pEFBos-Flag-STING vectors were obtained by inserting STING, eGFP and Flag sequences flanked by 5′ XhoI and 3′ NotI sites into the pEFBos vector. The pEFBos-based STING mutations (I10E, P11E, R14E, R14W, G15I, K20I, E38K, E38R, Y46R, L54D, E68Q, E69L, E69N, R76E, S80I, L93Y, L100E, Y106K, Y106R, W119C, W119K/M120K, A142D, C148E, E149F, N154S, A156G, H157K, I165H, R169A, R169K, R169W, D205H, V208P, P209K, D210P, M214K, L268I, G278F, F279V, R284S, Q287S, D301W, P317R, D319G, D319K/D320K, D320K, E339K, E340K, E339K/E340K, S366A and P371G) were obtained by site-directed mutagenesis. The pSIN-based STING and STING mutations (W119K/M120K, I165H and D319K/D320K) were generated by Gateway cloning by means of the pDONR 221 vector into the pSIN vector. The primers used for plasmid are provided in Supplementary Table 1.Generation of cell linesTo generate the IFN reporter cell line, the lentiviral plasmid41 (a gift from D. H. Raulet) encoding the tdTomato reporter gene driven by the IFN-stimulated response elements and the minimal mouse Ifnb promoter was transduced to HEK293T cells, followed by the selection under 100 μg ml−1 zeocin. For the eGFP-LC3B reporter, HEK293T cells were transduced with a pLVX lentiviral plasmid encoding eGFP-LC3B inserted between the 5′ XbaI and 3′ PmeI restriction sites, followed by fluorescence-activated cell sorting on the basis of eGFP signal.To establish HEK293T cells, BJ-5ta cells and THP-1 cells with inducible expression of distinct STING mutants, corresponding cells were transduced with a pSIN lentiviral vector carrying eGFP-STING or STING alone and a puromycin-resistance gene. Cells were selected with puromycin (1 μg ml−1 for HEK239T and BJ-5ta, 2.5 μg ml−1 for THP-1).Library design and constructionThe library design was adapted from ref. 60. In brief, the Homo sapiens STING sequence was (codon-optimized and) mutated systematically on each amino-acid position into all the other 19 types of amino acid. The full-length sequence was split into three segments: residues 1–155, 156–270 and 271–379. Within each segment, only the indicated residues were mutated whereas the remainder of the coding sequence remained WT. These three sublibrary oligonucleotide pools were purchased from Twist Biosciences and amplified individually using Phanta polymerase (Vazyme, P515-00). PCR products were then inserted into the pSIN vector through Gateway cloning.Library characterization and screeningSTING site-saturation mutagenesis library was transduced into HEK293T IFN or eGFP-LC3B reporter cell lines. Cells were then selected with puromycin (1 μg ml−1). Doxycycline (1 μg ml−1) was added 2 days before collection to induce the expression of STING variants. For the screen in presence of a ligand in IFN reporter cells, 100 nM cGAMP (Invivogen, tlrl-nacga23-1) (with 8 μg ml−1 digitonin (Thermo Fisher Scientific, BN2006)) was added 20 h before collection. For the screen with a ligand in eGFP-LC3B reporter cells, 2.5 μM cGAMP (with 8 μg ml−1 digitonin) was added 2 h before collection. After collection, eGFP-LC3B cells with STING mutagenesis libraries were resuspended in phosphate-buffered saline (PBS) containing 0.05% (w/v) saponin (Merck, 47036-50G-F) and incubated for 5 min to wash away cytosolic LC3B. Cells were then washed with PBS twice and incubated in PBS containing 1% (w/v) paraformaldehyde (PFA) (Thermo Fisher Scientific, J19943.K2) for 15 min for fixation. After centrifugation, cell pellets were resuspended in flow cytometry buffer (PBS containing 1% (w/v) bovine serum albumin (BSA) and 2 mM EDTA) for sorting on a Sony SH800 sorter. Cells were collected on the top 5–10% and the bottom 8–10% of the fluorescence signal (tdtomato for IFN reporter assay and eGFP for LC3B reporter assay). A portion before sorting was kept as the control.Next-generation sequencingGenomic DNA of cells collected in flow cytometry was extracted using QIAamp DNA Mini Kit (Qiagen, 51304) or QIAmp DNA blood midi kit (Qiagen, 51183) according to the manufacturer’s instructions. Illumina amplicons were generated by PCR of specific STING fragments using barcoded primers (Supplementary Table 1). These amplicons were purified using AMPure XP beads (Beckman, A63880) and sequenced using MiSeq (Illumina) pair-end 300 cycles method. A 30% high-diversity library was added to increase the sequencing accuracy.Next-generation sequencing data analysisThe quality of all reads was checked by FastQC (v.0.12.1) and filtered by Trimmomatic (v.0.39)62. Passing reads were merged using NGmerge tool (v.0.3)63. According to the designated reading frame, the merged reads were translated into amino-acid sequences and aligned to that of STING. The number of all aligned reads in a specific group G was denoted as Rtotal,G, within which the number of reads with the wild-type sequence RWT,G and the number of reads with a single amino-acid mutation Rsingle,G. Within the reads with single mutations, the raw count of each variant in a specific group G was denoted as Rij,G, where i is the position of the mutation (1, 2, …, 379) and j is the type of the mutation (A, C, …, Y). To avoid bias resulting from too few counts, the threshold of the raw count of each single mutation was set to 20. The functional score for each single amino acid mutation in one screening set up was defined as the enrichment of this mutant in the top group compared with the control group after normalizing to the enrichment of the wild-type sequence:$${e}_{ij}={\log }_{2}\frac{{R}_{ij,{\rm{t}}{\rm{o}}{\rm{p}}}/{R}_{{\rm{s}}{\rm{i}}{\rm{n}}{\rm{g}}{\rm{l}}{\rm{e}},{\rm{t}}{\rm{o}}{\rm{p}}}}{{R}_{ij,{\rm{c}}{\rm{o}}{\rm{n}}{\rm{t}}{\rm{r}}{\rm{o}}{\rm{l}}}/{R}_{{\rm{s}}{\rm{i}}{\rm{n}}{\rm{g}}{\rm{l}}{\rm{e}},{\rm{c}}{\rm{o}}{\rm{n}}{\rm{t}}{\rm{r}}{\rm{o}}{\rm{l}}}}-{\log }_{2}\frac{{R}_{{\rm{W}}{\rm{T}},{\rm{t}}{\rm{o}}{\rm{p}}}/{R}_{{\rm{t}}{\rm{o}}{\rm{t}}{\rm{a}}{\rm{l}},{\rm{t}}{\rm{o}}{\rm{p}}}}{{R}_{{\rm{W}}{\rm{T}},{\rm{c}}{\rm{o}}{\rm{n}}{\rm{t}}{\rm{r}}{\rm{o}}{\rm{l}}}/{R}_{{\rm{t}}{\rm{o}}{\rm{t}}{\rm{a}}{\rm{l}},{\rm{c}}{\rm{o}}{\rm{n}}{\rm{t}}{\rm{r}}{\rm{o}}{\rm{l}}}},$$$${\rm{for}}\,i=1,2,\ldots ,379,\,j\in \{{\rm{A}},{\rm{C}},\ldots ,{\rm{Y}}\},{R}_{{ij},{\rm{top}}} > 20,{R}_{{ij},{\rm{ctrl}}} > 20$$TransfectionCells plated in a 12-well plate were transfected with plasmids using GeneJuice transfection reagent (Millipore, 70967) following the manufacturer’s protocol.Flow cytometryCells were collected, washed with PBS twice and fixed with 1% PFA for 15 min at room temperature. After washing with PBS twice, cells were resuspended in flow cytometry buffer (PBS containing 1% (w/v) BSA and 2 mM DETA) and loaded onto a BD LSRFortessa Cell Analyzer. For eGFP-LC3B reporter assays, cells were incubated with 0.05% Saponin in PBS for 5 min at room temperature before the fixation. Data were analysed with FlowJo (v.10.10.0).Quantitative PCRCells were lysed and the total RNA was extracted using the RNeasy Mini Kit (Qiagen, 74104) according to the manufacturer’s instructions. Complementary DNA was synthesized using the PrimeScript RT Reagent Kit (Takara, RR047B), and quantitative PCR (qPCR) was performed using the Maxima SYBR Green/ROX qPCR Master Mix (Thermo Fisher Scientific, K0223) on a QuantStudio 7 Real-Time PCR System (Thermo Fisher Scientific). Gene expression was normalized to glyceraldehyde 3-phosphate dehydrogenase. Primer sequences are listed in Supplementary Table 1.ELISATHP-1 STING1 knockout cells reconstituted with eGFP-tagged wild-type or mutant STING were treated 100 ng ml−1 GM-CSF (Miltenyi Biotec, 130-095-372) and 100 ng ml−1 IL-4 (Miltenyi Biotec, 130-093-917) for 5 days to induce differentiation into dendritic cell-like cells. After a 3-day resting period, doxycycline was added to induce expression of wild-type or mutant STING for 24 h. Supernatants were collected and centrifuged to remove cell debris and dead cells. IL-8 concentrations were quantified by enzyme-linked immunosorbent assay (ELISA) using the BD OptEIA Human IL-8 ELISA Set (BD Biosciences, 555244) according to the manufacturer’s instructions.ImmunoblottingCells were collected, quickly rinsed with 1× PBS, then centrifuged at 2,000 rpm for 5 min. Pellets were boiled with 1× loading buffer (4×, 200 mM Tris pH 6.8, 8% SDS, 40% glycerol, 0.4 M dithiothreitol, 0.4% (w/v) bromophenol blue) for 10 min. Proteins were resolved by SDS–PAGE using SurePAGE precast gels (GenScript) and transferred to nitrocellulose membranes using the Trans-Blot Turbo RTA Midi Nitrocellulose Transfer Kit (Bio-Rad) following the manufacturer’s instructions. Membranes were blocked with 5% (w/v) skim milk in PBS with Tween (PBST) (PBS + 0.05% (v/v) Tween-20) at room temperature for 1 h and then incubated with the primary antibody (diluted in 5% (w/v) BSA in PBST) at 4 °C overnight. After washing in PBST, membranes were incubated with the secondary antibody (diluted in 5% (w/v) BSA in PBST) at room temperature for 1 h. Membranes were washed with PBST, visualized with Western Blotting Detection Reagent (Bio-Rad) and imaged with using the ChemiDoc XRS Biorad Imager and Image Lab Software. A list of antibodies is provided in Supplementary Table 2.ImmunofluorescenceCells were seeded at 10,000 cells per well in CellCarrier-96 Ultra plates (PerkinElmer, 6055302) and either transfected with STING variants or induced to express STING variants with doxycycline for 24 h. Cells were then washed twice with PBS and fixed in 4% (w/v) PFA in CBS buffer (10 mM MES pH 6.9, 138 mM KCl, 2 mM MgCl2, 2 mM EGTA) for 5–10 min at room temperature. Following three 5-min PBS washes, cells were permeabilized and blocked for 1–2 h in PBS containing 0.1% (w/v) saponin and 5% (v/v) heat-inactivated FBS, then incubated overnight at 4 °C with primary antibodies diluted in staining buffer (PBS with 0.1% (w/v) saponin and 1% (w/v) BSA). Antibodies and dilutions are listed in Supplementary Table 2. After three 5-min PBS washes, cells were incubated with secondary antibodies for 1 h at room temperature in the dark, followed by 2 more PBS washes and nuclear counterstaining with Hoechst 33342 (0.2 µg ml−1, 20 min). Imaging was performed on a Zeiss LSM 980 inverted confocal microscope using a Plan-Apochromat ×63/1.40 oil objective. Image analysis and quantification were performed in Fiji (v.2.16.0)64.Bulk RNA-seqTHP-1 STING1 knockout cells reconstituted with eGFP-tagged wild-type or mutant STING were treated with doxycycline for 24 h and the RNA was isolated with RNeasy micro kit (Qiagen, 74004) according to the manufacturer’s instructions. RNA was further processed for sequencing by the Gene Expression Core Facility at EPFL. In brief, samples were prepared with NEBNext Ultra II Directional RNA Library Prep Kit (New England Biolabs, E7760L) with ribodepletion starting from 750 ng of RNAs. The quality control was performed using Qubit DNA HS and TapeStation 4200 (Agilent). Libraries bearing unique indexes were loaded onto the Aviti system (Element Biosciences) sequencing with the 75 cycles (pair-end) method.Bulk RNA-seq analysisThe analysis of the RNA-seq was performed in the Galaxy server (https://usegalaxy.eu/)65. In brief, RNA was mapped to human genome assembly GRCh38 (hg38.ensGene.gtf.gz) using STAR aligner (v.2.7.11b)66 and the counts were generated with featureCounts (v.2.1.1)67. Differential gene expression analysis was done in DESeq2 (v.2.11.40.8)68. Heatmaps were generated using the Python package matplotlib (v.3.10.0)69.Organelle pH sensor reporter assayHEK293T cells were transduced with lentiviral SEP/mRuby3 ratiometric reporters targeted to MGAT (alpha-1,3-mannosyl-glycoprotein 2-beta-N-acetylglucosaminyltransferase) (Addgene, 200937) or GALT (galactose-1-phosphate uridylyltransferase) (Addgene, 200938), in which SEP and mRuby3 were used as pH-sensitive and reference fluorescent proteinsm respectively, as previously reported in ref. 12. Transduced cells were sorted based on mRuby3 expression by means of BD FACSAria III Cell Sorter, referred to as pH sensor reporter cells. Wild-type STING or variants were transfected into pH sensor reporter cells for 24 h. On collection, cells were wash with PBS twice, resuspended in flow cytometry buffer (PBS containing 1% (w/v) BSA and 2 mM DETA) and loaded onto BD LSRFortessa Cell Analyzer. The SEP/mRuby3 intensity of each cell was calculated and plotted in GraphPad.Protein expression and purificationFlag-tagged human wild-type STING and Flag-tagged STING mutants in the pEFBos vector were expressed in HEK293 cells. Cells were transfected at a density of 2.0 × 106 cells per ml in RPMI 1640 medium (Thermo Fisher Scientific/Life Technologies) supplemented with 0.1% Pluronic F-68 (BioChemica). Sterile plasmid DNA was added at a final concentration of 1.5 mg per litre, mixed by gentle shaking, followed by the addition of PEI-Max (1 mg ml−1 stock; Polysciences, Chemie Brunschwig) to a final concentration of 3 mg per litre of transfection. The mixture was incubated for 1.5 h at 37 °C with stirring.After transfection, the culture was diluted to 1.0 × 106 cells per ml in HyCell TransFx-H medium (Cytiva) supplemented with valproic acid (Cayman, Chemie Brunschwig) to a final concentration of 3.75 mM and incubated at 37 °C for 3 days with continuous stirring. Cells were then collected by centrifugation, and the supernatant was discarded. The cell pellet was washed once with cold 1× PBS, centrifuged again and the resulting pellet was frozen immediately on dry ice for storage.Cells were resuspended in cold buffer containing 20 mM Tris-HCl (pH 7.5), 150 mM NaCl and cOmplet protease inhibitor (Roche). The suspension was disrupted by sonication, and cell debris was removed by centrifugation at 5,000g for 15 min at 4 °C. Membranes were subsequently pelleted by ultracentrifugation at 100,000g for 1 h at 4 °C. Membranes containing STING or its mutants were solubilized in 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% (w/v) lauryl maltose neopentyl glycol (LMNG) (Anatrace) and 0.1% (w/v) cholesteryl hemisuccinate (CHS) (Anatrace) for 2 h at 4 °C. The solubilized material was clarified by centrifugation, and the supernatant was incubated overnight with anti-Flag M2 affinity gel (Sigma, A2220-5ML) at 4 °C. The resin was washed with 20 mM Tris (pH 7.5), 150 mM NaCl and 0.01% (w/v) LMNG/0.001% (w/v) CHS, and bound proteins were eluted with the same buffer supplemented with 0.2 mg ml−1 3× Flag peptide (Sigma, F4799-4MG). All mutants were generated using PCR-based mutagenesis with appropriate primers and verified by DNA sequencing.Cryo-EM data acquisitionFlag-tagged STING and mutant proteins eluted from the Flag resin were concentrated to 1 mg ml−1 in buffer containing 20 mM Tris (pH 7.5), 150 mM NaCl, 0.01% LMNG and 0.001% CHS. A 500-µl aliquot of the sample was injected onto a Superdex 200 Increase 10/300 GL column (Cytiva) equilibrated with 20 mM Tris (pH 7.5), 150 mM NaCl, 0.001% LMNG, 0.0001% CHS and 0.0003% glyco-diosgenin.To comprehensively characterize the oligomeric state, samples without size-exclusion chromatography purification were applied directly onto glow-discharged holey carbon grids (Q250AR1.3, Quantifoil, Au, R 1.2/1.3, 300 mesh, Electron Microscopy Sciences). Grids were blotted for 4 s and plunge-frozen in liquid ethane using a Vitrobot (Thermo Fisher Scientific) operated at 4 °C and 100% humidity.Cryo-EM data were collected on a Thermo Fisher Scientific Titan Krios G4 transmission electron microscope equipped with a Cold-FEG and a Falcon IV detector operating in electron-counting mode. Falcon IV gain references were acquired immediately before data collection. Automated data acquisition was performed using EPU v.2.12.1 with the aberration-free image shift protocol, recording four micrographs per ice hole. Videos were collected at a nominal magnification of ×96,000, corresponding to a pixel size of 0.83 Å at the specimen level, with a defocus range of −0.8 µm to −1.8 µm. The total electron dose was set to 50 e− Å−2, with an exposure time of roughly 5 s per video. Information detailing cryo-EM data collection, refinement and validation statistics is provided in Supplementary Table 4.Cryo-EM data processingMotion correction of raw micrograph stacks was performed without binning using the cryoSPARC (v.4.7.1)70 implementation of motion correction. Particles were automatically picked using blob picking and initially extracted with a fourfold downsampling factor. Several rounds of two-dimensional classification were carried out to remove poorly defined particles. The selected high-quality particles were used for ab initio reconstruction with one to three classes, followed by heterogeneous refinement to isolate the subset corresponding to STING. These particles were then re-centred, re-extracted and subjected to non-uniform refinement in cryoSPARC. Reported resolutions are based on the gold-standard Fourier shell correlation 0.143 criterion, and local resolution variations were estimated within cryoSPARC.Model building and refinementAn initial atomic model was generated using ModelAngelo based on the cryo-EM density map71. Manual model adjustments, including side-chain fitting and loop rebuilding, were performed in Coot and further optimized interactively in ISOLDE72,73. The refined models were subsequently subjected to real-space refinement using Phenix, with secondary structure restraints and non-crystallographic symmetry constraints applied where appropriate74. Model quality was assessed throughout the process using geometry validation tools implemented in Phenix.Human genetic STING1 variant and clinical presentationThe proband was a 34-year-old man of North African origin. He presented at age 32 years with dyspnoea, chronic cough and skin lesions. Chest computed tomography demonstrated features of interstitial lung disease with ground-glass opacities, traction bronchiectasis and peribronchovascular opacities (Extended Data Fig. 10e). Skin capillaroscopy revealed megacapillaries and haemorrhages suggestive of scleroderma. Antinuclear antibodies were positive at a titre of 1:1,280 with a cytoplasmic fluorescence pattern, as were anti-TRIM21 and anti-EIF2B antibodies. A six-ISG signature was elevated on three separate occasions over a period of 12 months. Targeted next-generation sequencing filtered against an interstitial lung disease gene panel revealed a heterozygous variant in STING1 (NM_198282.4):c.956A>G, p.(Asp319Gly) (Extended Data Fig. 10f). This variant is absent from gnomAD v.4.1.0, and is predicted to be non-deleterious on the basis of in silico pathogenicity scores (CADD v.21.9, SIFT tolerated, Polyphen-2 HumVar benign, AlphaMissense likely benign, REVEL benign). The variant was inherited from the patient’s mother who reported no lung or skin problems, but had a diagnosis of Budd–Chiari syndrome leading to liver transplantation at the age of 34 years. The translational study was approved by the Comité de Protection des Personnes (ID-RCB/EUDRACT: 2014-A01017-40; revalidated in 2022 and 2025). Written informed consent was obtained from the patient.Multiple-sequence alignmentA total of 184 vertebrate STING orthologue sequences were retrieved from the Ensembl Genome Browser. Multiple-sequence alignment was performed using MAFFT in SnapGene, and the resulting alignment was used for BLOSUM62-based conservation analysis and the generation of sequence logos.Clinical impact analysisPublicly available databases used in this study include ClinVar that is available through the National Institutes of Health at https://www.ncbi.nlm.nih.gov/clinvar/ (with the gene query STING1); gnomAD available through the Broad Institute at https://gnomad.broadinstitute.org/ (version 4.1, with the gene query TMEM173) and COSMIC available through the Sanger Institute at https://cancer.sanger.ac.uk/cosmic (version 102, with the gene query TMEM173). Figures were prepared with GraphPad.Statistics and data reproducibilityFor all experiments, n = 3 biological replicates were performed unless otherwise noted. P values were calculated using one-way analysis of variance (ANOVA) followed by Tukey’s multiple-comparison test and ordinary two-way ANOVA unless otherwise indicated.Reporting summaryFurther information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
The mutational landscape of STING-induced immunity - Nature
A massively parallel assay systematically charts the sequence-function landscape of the STING signalling protein, and the findings define molecular principles that tune STING activity and show its functional potential across immune contexts.
13,503 words~61 min read