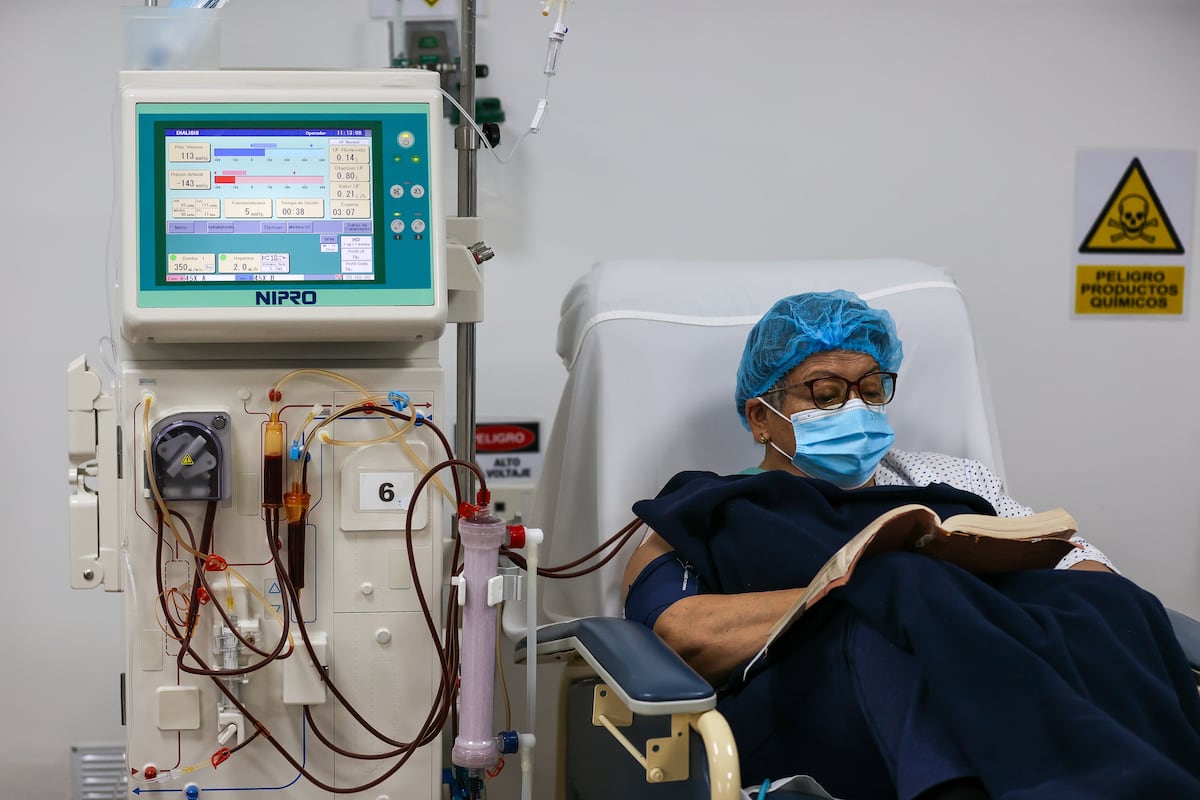
Aunque suelen relacionarse con la dieta, la falta de hidratación o ciertos hábitos de vida, los cálculos renales no siempre responden a estas causas. En determinados casos ⎯sobre todo en niños y en adultos que presentan episodios recurrentes⎯ pueden ser indicio de una patología genética poco frecuente: la hiperoxaluria primaria tipo 1 (HP1). Ralentizar la progresión de la enfermedad y ayudar a preservar la función renal es el reto de compañías como Anlylam Pharmaceuticals
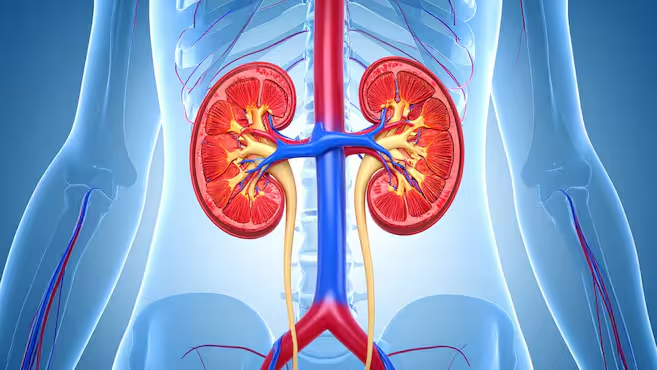
La hiperoxaluria primaria es un grupo de enfermedades hereditarias muy poco frecuentes que afectan al metabolismo hepático; pueden manifestarse en cualquier momento de la vida, desde la infancia hasta la edad adulta. La forma más habitual es la hiperoxaluria primaria tipo 1 (HP1). Representa aproximadamente entre el 70 y el 80 % de los casos y se debe a mutaciones en el gen AGXT, responsable de la enzima hepática alanina: glioxilato aminotransferasa, cuya alteración provoca una producción excesiva de oxalato.
Cuando el oxalato se acumula, se combina con el calcio y forma cristales que se depositan en el riñón, lo que favorece la aparición de cálculos renales y nefrocalcinosis (depósitos de calcio en el tejido renal). La manifestación más habitual de la HP1 son los episodios recurrentes de litiasis renal. La complicación más grave es el deterioro progresivo de la función renal, que puede avanzar hasta una insuficiencia terminal e incluso resultar mortal.