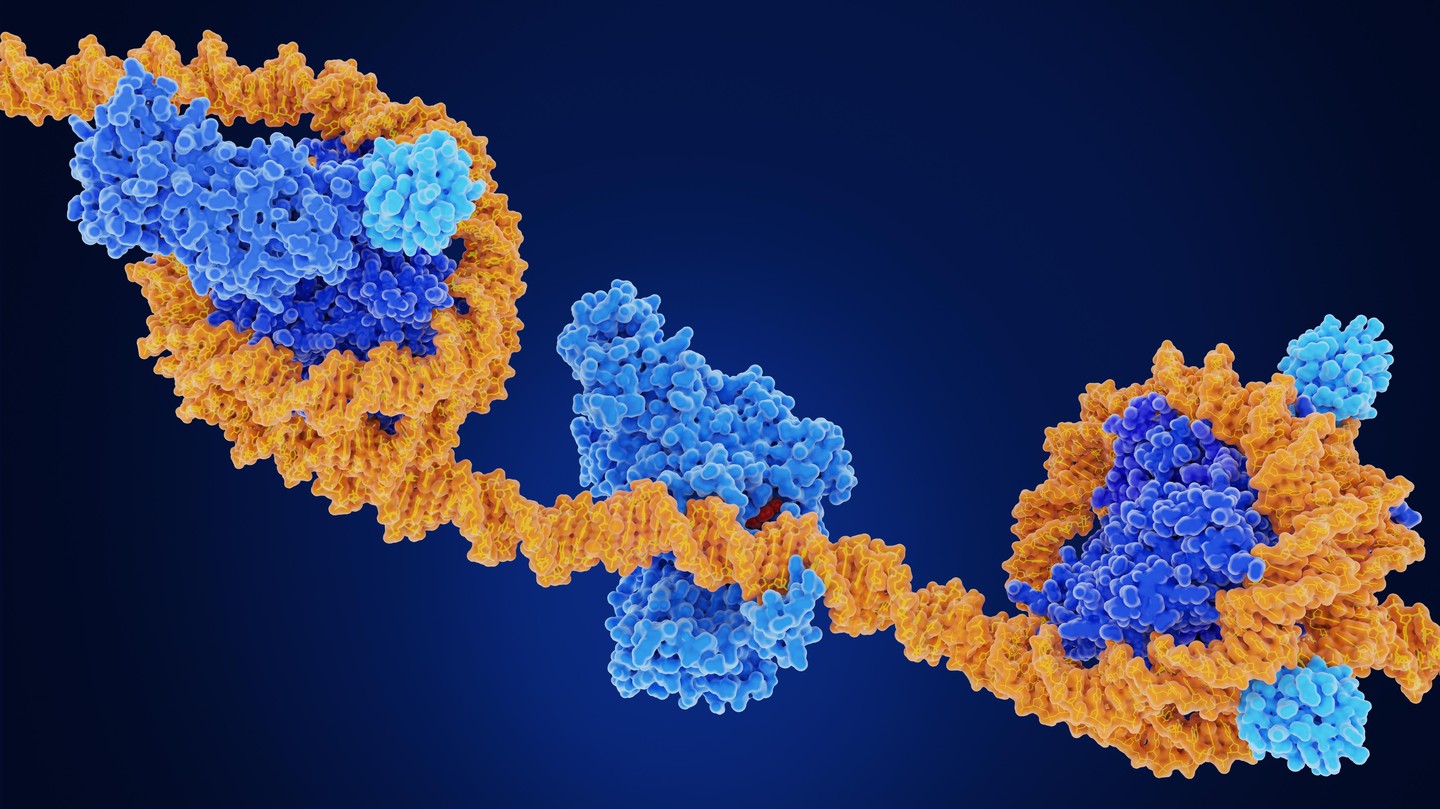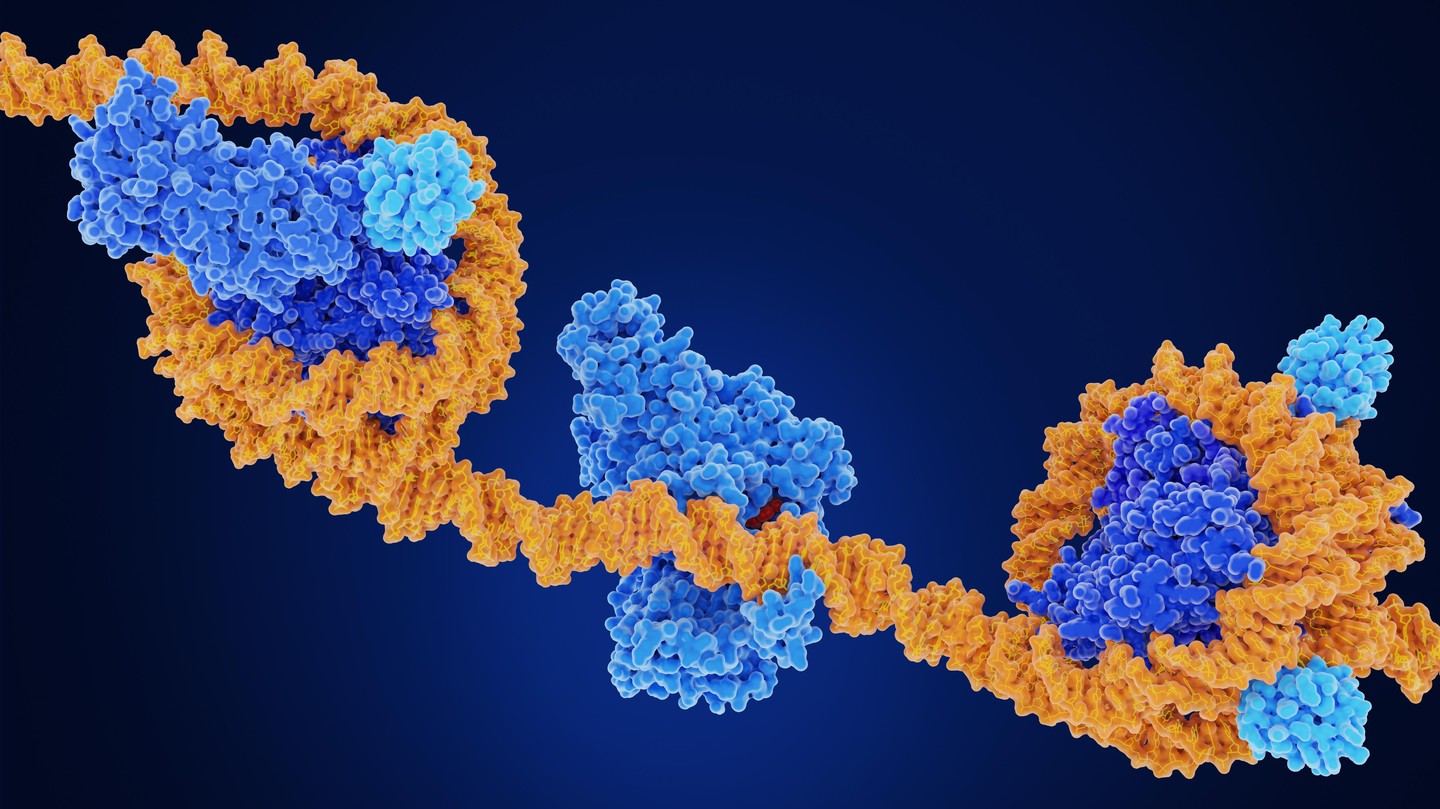
Des chercheur·euses de l’EPFL ont mis au point une méthode rapide, évolutive et économique permettant de cartographier la méthylation de l’ADN dans les cellules, les organoïdes et les tumeurs cérébrales, avec des besoins en séquençage fortement réduits par rapport aux méthodes de référence actuelles.L’ADN ne contient pas seulement des gènes. Des marqueurs chimiques fixés à l’ADN contribuent également à réguler l’activation ou la désactivation des gènes. L'une des plus importantes de ces marques est la méthylation de l'ADN, une marque moléculaire liée au développement, au vieillissement et à des maladies telles que le cancer.Les scientifiques disposent déjà de moyens pour mesurer la méthylation de l'ADN à l'échelle du génome, mais le problème réside dans le coût : la référence actuelle, le séquençage bisulfite du génome entier, nécessite d'énormes quantités de données de séquençage pour obtenir des résultats précis, ce qui rend les études à grande échelle coûteuses et difficiles à mener.Les chercheur·euses ont passé des années à chercher des alternatives rentables. Certaines méthodes ne s’intéressent qu’à des parties sélectionnées du génome. D’autres enrichissent l’ADN méthylé à l’aide d’anticorps ou de protéines de liaison.Ces approches réduisent les coûts, mais sacrifient souvent la résolution, la sensibilité ou la flexibilité. Beaucoup nécessitent également de grandes quantités de matériel biologique, ce qui les rend difficiles à utiliser sur des échantillons rares ou dans les méthodes émergentes à l’échelle de la cellule unique.Transformer Tn5 en détecteur de méthylationUne équipe dirigée par Fides Zenk à l’EPFL a désormais mis au point une nouvelle méthode appelée CmeCUT&Tag qui établit le profil de la méthylation de l’ADN à l’échelle du génome en utilisant bien moins de séquençage que les approches standard.Ces travaux montrent que la méthode permet de détecter des profils de méthylation dans des cellules souches, des organoïdes cérébraux, des embryons de poisson-zèbre et des tumeurs cérébrales humaines, tout en réduisant les besoins en séquençage de 10 à 40 fois.Les chercheur·euses ont mis au point cette méthode en combinant Tn5, une enzyme largement utilisée dans le séquençage du génome, avec des domaines protéiques qui se lient à l’ADN méthylé. Cela dirige l’enzyme vers les régions méthylées, ce qui permet aux chercheur·euses de séquencer principalement ces parties du génome au lieu de tout séquencer.La version la plus performante correspondait étroitement aux régions hautement méthylées détectées par les résultats standard du séquençage bisulfite du génome entier, tout en utilisant beaucoup moins de données de séquençage. La méthode a également fonctionné dans des noyaux cellulaires intacts et de l'ADN isolé, y compris à partir de faibles quantités d'échantillons.Validation de la méthode Pour confirmer que la méthode détecte bien la méthylation de l'ADN, les chercheur·euses ont chimiquement réduit les niveaux de méthylation de l'ADN dans des cellules souches à l'aide d'un médicament bloquant l'enzyme responsable du maintien de ces marques de méthylation. Les signaux émis par CmeCUT&Tag ont chuté brutalement après le traitement, démontrant que la méthode suit spécifiquement l'ADN méthylé.L'équipe a également adapté l'approche pour obtenir une résolution au niveau des paires de bases lorsque cela était nécessaire. En combinant CmeCUT&Tag avec des méthodes de conversion au bisulfite ou enzymatique, ils ont pu identifier les cytosines méthylées avec une précision au niveau du nucléotide tout en ne séquençant que les régions ciblées.Les chercheur·euses estiment que le séquençage au bisulfite traditionnel du génome entier nécessite environ 800 millions à 1 milliard de paires de lectures pour couvrir le génome humain à une profondeur standard. En revanche, CmeCUT&Tag, combiné à des méthodes de conversion, a permis de réduire ce nombre à environ 20 à 100 millions de lectures.Du développement cérébral au cancerL'étude a également exploré des applications biologiques et cliniques. Dans des organoïdes cérébraux humains cultivés à partir de cellules souches pluripotentes induites, les chercheur·euses ont suivi l'évolution des profils de méthylation au cours du développement neural. Ils ont observé des gains et des pertes de méthylation dans des gènes liés au développement des neurones, à la prolifération cellulaire et aux processus embryonnaires.L'équipe a également testé si la méthode pouvait classer les tumeurs cérébrales à l'aide des profils de méthylation de l'ADN, un outil de plus en plus important dans le diagnostic du cancer. Elle a analysé 24 biopsies de tumeurs cérébrales chez l'adulte et a correctement attribué la famille de classe de méthylation à 17 des 19 tumeurs qui répondaient aux critères de filtrage.Enfin, les chercheur·euses ont montré que la méthode fonctionne sur des embryons de poisson-zèbre, ce qui suggère qu'elle peut être appliquée à différentes espèces.La méthode présente toutefois encore des limites. Dans sa forme actuelle, elle ne permet pas de distinguer deux versions chimiques étroitement apparentées de l'ADN méthylé, et elle donne les meilleurs résultats dans les régions présentant des niveaux de méthylation relativement élevés. L'adaptation de la cartographie de la méthylation de l'ADN unicellulaire à l'aide de ce protocole est déjà possible avec des plateformes à base de gouttelettes, mais nécessitera une caractérisation plus poussée pour distinguer des états épigénétiques étroitement apparentés.Néanmoins, les chercheur·eusesestiment que le coût réduit et la flexibilité de CmeCUT&Tag pourraient rendre l'analyse de la méthylation de l'ADN à l'échelle du génome plus accessible pour des études de recherche à plus grande échelle et de futures applications cliniques.Autres contributeursHôpital universitaire de ZurichFinancementFonds national suisse (FNS)Programme de financement « Innovate for Life » de l'EPFLFondation NeuroNARéférencesHanrong Hu, Nahuel Simonet, Ece Naz Bilgiç, Heather Murray, Regina Reimann, Markus Rechsteiner, Fides Zenk. A scalable Tn5-based method for genome-wide DNA methylation profiling in development and disease. Nature Communications 22 mai 2026. DOI: 10.1038/s41467-026-73325-4
Une approche scalable et économique pour lire la méthylation de l'ADN
Des chercheur·euses de l’EPFL ont mis au point une méthode rapide, évolutive et économique permettant de cartographier la méthylation de l’ADN dans les cellules, les organoïdes et les tumeurs cérébrales, avec des besoins en séquençage fortement réduits par rapport aux méthodes de référence actuelles.
904 words~4 min read