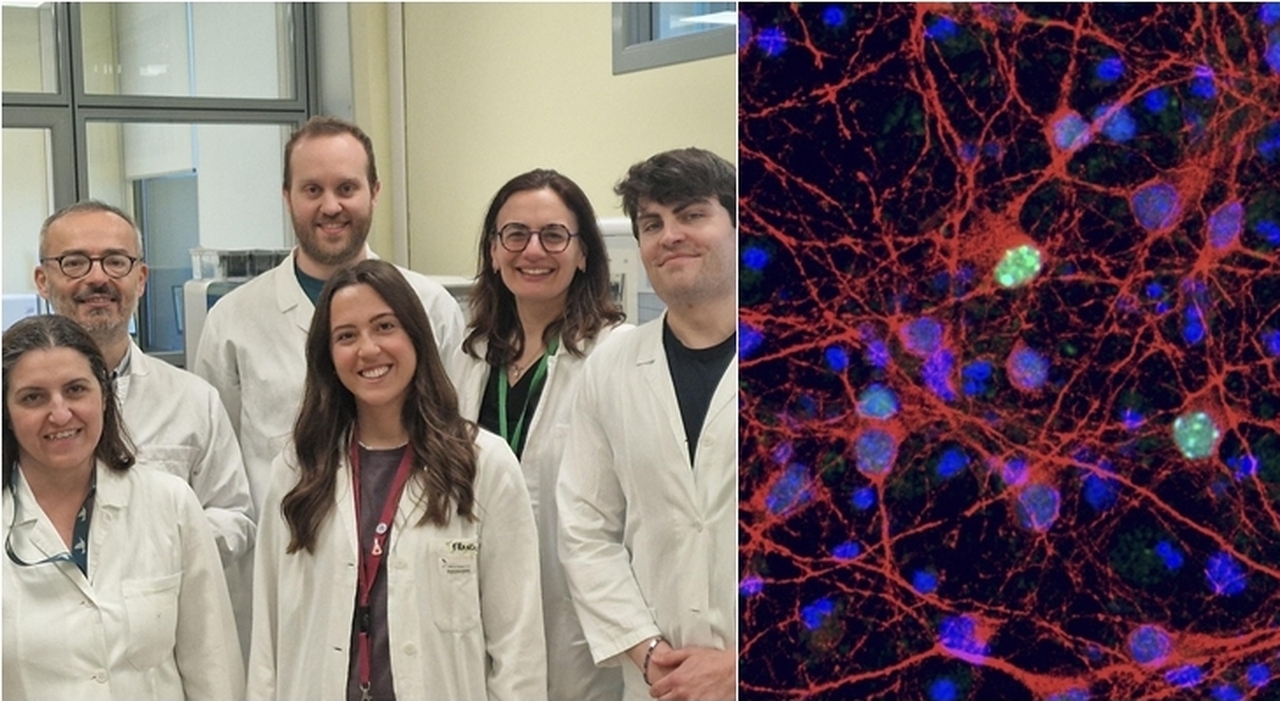
Una nuova ricerca italiana ridefinisce in profondità il ruolo della proteina MeCP2 nella sindrome di Sindrome di Rett, offrendo prospettive inedite per lo sviluppo di terapie mirate. La patologia, che colpisce prevalentemente le bambine, è causata dalla perdita di funzione del gene MECP2 ed è caratterizzata da una progressiva regressione delle capacità motorie, del linguaggio e dell’interazione sociale. Nonostante decenni di studi, non esistono ancora trattamenti in grado di arrestarne o invertirne il decorso. Il lavoro, condotto da un gruppo di ricerca dell’Consiglio Nazionale delle Ricerche – in particolare dall’Istituto di neuroscienze (Cnr-In) e dall’Istituto di tecnologie biomediche (Cnr-Itb) di Milano – in collaborazione con l’IRCCS Ospedale San Raffaele, è stato pubblicato su Nature Communications e introduce un cambio di paradigma nella comprensione della funzione di MeCP2.
Tradizionalmente, la proteina MeCP2 è stata considerata principalmente come un repressore trascrizionale, ovvero un regolatore capace di “spegnere” l’espressione genica. Lo studio guidato da Vania Broccoli e Mirko Luoni dimostra invece che MeCP2 svolge anche una funzione attiva nel promuovere l’espressione di geni fondamentali per lo sviluppo neuronale. In particolare, la proteina agisce in cooperazione con il complesso epigenetico SWI/SNF, un sistema molecolare che modula l’accessibilità del Dna e quindi la possibilità per i geni di essere trascritti.