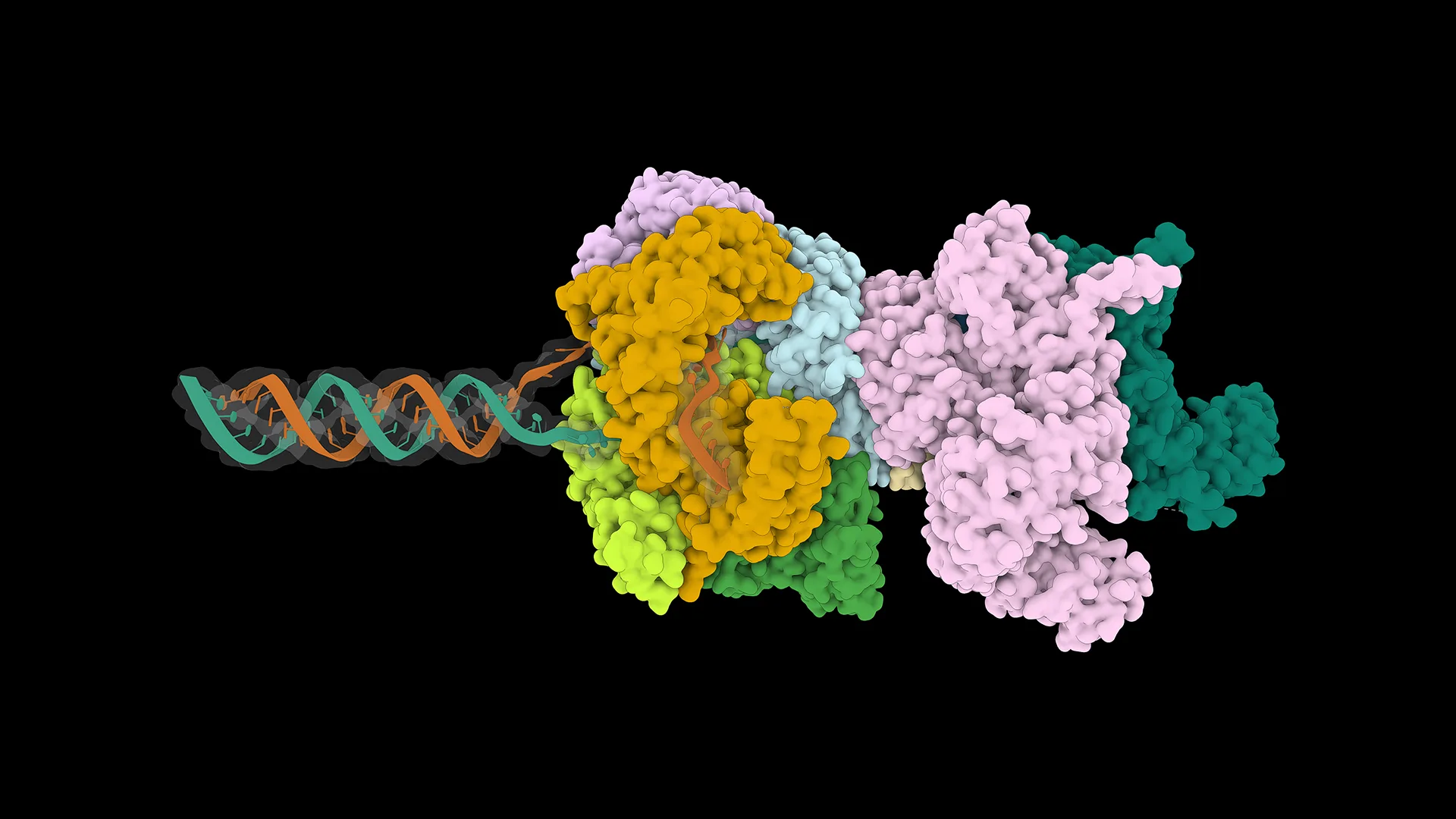
Biomolecular structure prediction and co-folding with models like OpenFold3 are now mainstream, large-scale workloads powering drug discovery and protein design. Increasingly, they’re driven end-to-end by AI agents. For an agent to run that pipeline well, every step needs to be fast and scalable: Multiple Sequence Alignment (MSA) generation, co-folding inference, serving, and multi-GPU scale-out. A bottleneck anywhere limits overall throughput.
Speed and memory-efficiency are critical for key drug discovery workflows such as virtual screening and prediction of large molecular assemblies. In virtual screening, millions to billions of compounds are screened against one or a few protein targets. While co-folding models often give the best predicted structures, they can be expensive to run, making them impractical for virtual screening applications. That is where NVIDIA acceleration becomes key, making possible the deployment of OpenFold3 and related methods at the scale of large compound libraries.
Speed is also important for predicting large molecular assemblies involving multiple proteins and thousands of amino acid residues, as co-folding model runtime scales cubically with the number of residues. An even bigger challenge, however, is memory use, as single GPU memory can be limited, placing a hard ceiling on the size of complexes that can be predicted in one shot. Methods to reduce memory requirements, and that distribute prediction tasks across multiple GPUs, would enable qualitatively new applications that are simply unfeasible today.