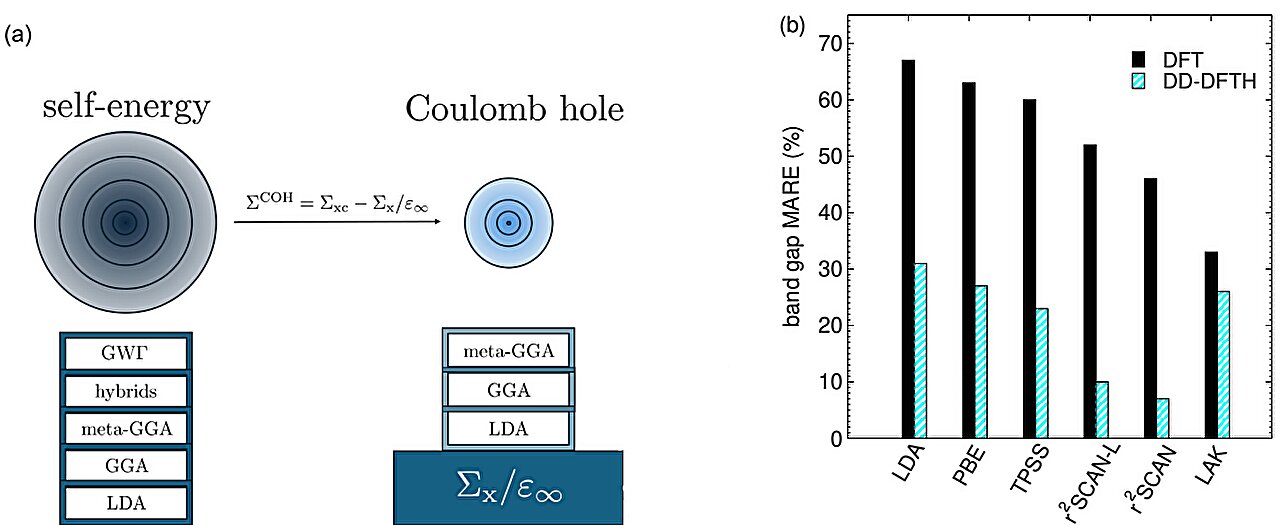
Une étude menée par des chercheurs de l’EPFL présente une nouvelle méthode de calcul qui prédit les propriétés électroniques des semi-conducteurs avec plus de précision, y compris des matériaux que les méthodes existantes peuvent parfois classer à tort comme des métaux.Les semi-conducteurs occupent une place centrale dans la technologie moderne. Ils sont utilisés dans les puces informatiques, les cellules solaires, les capteurs, les LEDs et les dispositifs de communication. Avant de fabriquer de nouveaux matériaux semiconducteurs en laboratoire, les chercheurs les testent souvent au préalable par des simulations de mécanique quantique.L’un des principaux outils utilisés dans ce domaine est la théorie de la fonctionnelle de la densité, ou DFT, une méthode de modélisation numérique qui, plutôt que de suivre chaque électron individuellement, utilise la densité globale de leur nuage pour calculer rapidement la structure atomique et l’énergie d’un matériau.La DFT offre aux chercheurs un moyen pratique de calculer le comportement des électrons dans les matériaux et est largement utilisée car elle répresente un bon équilibre entre précision et coût de calcul. Toutefois, elle présente une faiblesse persistante : elle sous-estime généralement la largeur de la bande interdite du matériau.La bande interdite est la différence d’énergie qui détermine avec quelle facilité les électrons se déplacent dans un matériau. Elle permet de déterminer si un matériau se comporte comme un métal, un semi-conducteur ou un isolant. Elle influe également sur la façon dont un matériau absorbe la lumière. Cela est important pour des technologies telles que les cellules solaires, les photodétecteurs et d’autres dispositifs électroniques.Le problème devient plus grave pour les semi-conducteurs à faible bande interdite. Ces matériaux sont importants pour l’optronique, la thermoélectrique et la spintronique. Les méthodes DFT standard peuvent prédire à tort que certains d’entre eux se comportent comme des métaux, ce qui peut orienter les chercheurs dans la mauvaise direction lors du criblage de nouveaux matériaux.Une nouvelle méthode : DD-r²SCANHUne équipe de chercheurs de l’EPFL, Stefan Riemelmoser, Xun Xu et Alfredo Pasquarello, a développé une méthode appelée DD-r²SCANH pour résoudre ce problème.L’innovation de DD-r²SCANH réside dans la combinaison de deux outils mathématiques : premièrement, une fonctionnelle de densité moderne appelée r²SCAN, qui est une formule mathématique permettant de prédire rapidement les interactions électroniques locales sans avoir à examiner l’ensemble de la densité électronique du matériau ; deuxièmement, un algorithme intelligent qui ajuste automatiquement la formule r²SCAN en y intégrant des interactions électron-électron à longue portée.Dans ces “fonctionnelles hybrides dépendantes du diélectrique”, la quantité de mélange dépend de la constante diélectrique du matériau, qui décrit la force de réponse du matériau à un champ électrique. Si cette valeur est erronée, la prédiction finale peut l’être aussi.De meilleures constantes diélectriques et des prédictions plus précises de la bande interditeLes chercheurs ont constaté que r²SCAN prédit les constantes diélectriques avec une précision bien supérieure à celle de la fonctionnelle PBE, la fonctionnelle de densité la plus largement utilisée en physique et en science des matériaux pour prédire la liaison atomique et le comportement des matériaux.Sur 39 semi-conducteurs et isolants bien caractérisés, r²SCAN a réduit l’erreur moyenne sur les constantes diélectriques de 26 % à 9 %.Ces améliorations de la structure électronique sont également héritées par les fonctionnelles hybrides dépendantes du diélectrique. DD-r²SCANH a atteint une erreur moyenne sur la bande interdite de 7 %, contre 27 % pour la fonctionnelle hybride dépendante du diélectrique basée sur PBE standard. Elle a particulièrement bien fonctionné pour les semi-conducteurs à faible bande interdite difficiles comme le germanium et l’arseniure d’indium, pour lesquels PBE peut prédire un faux état métallique.Performance sur de multiples propriétésL’équipe a également testé d’autres propriétés, notamment les potentiels d’ionisation, les masses effectives et les structures cristallines. DD-r²SCANH a amélioré les prédictions des potentiels d’ionisation tout en maintenant une bonne précision pour les autres propriétés. Ses performances se sont rapprochées de celles des méthodes avançées à plusieurs corps, qui sont très précises mais bien plus coûteuses en calcul.Ce travail fournit aux scientifiques des matériaux un outil plus fiable pour prédire les propriétés des semi-conducteurs. Étant donné que r²SCAN prédit avec précision les constantes diélectriques, le paramètre de mélange dans DD-r²SCANH peut être déterminé à partir des premiers principes. Cela signifie que la méthode peut également être appliquée à la découverte de matériaux. Elle pourrait aider les chercheurs à cribler les matériaux plus efficacement pour les futures technologies électroniques et énergétiques.Autres contributeursBeijing Computational Science Research CenterFinancementCSEA-EPFL, CSCSRéférencesStefan Riemelmoser, Xun Xu, Alfredo Pasquarello. Dielectric-dependent hybrid functional based on meta-GGA. Nature Communications, 04 juillet 2026. DOI: 10.1038/s41467-026-75146-x
Une meilleure prédiction des propriétés des semi-conducteurs
Une étude menée par des chercheurs de l’EPFL présente une nouvelle méthode de calcul qui prédit les propriétés électroniques des semi-conducteurs avec plus de précision, y compris des matériaux que les méthodes existantes peuvent parfois classer à tort comme des métaux.
742 words~3 min read