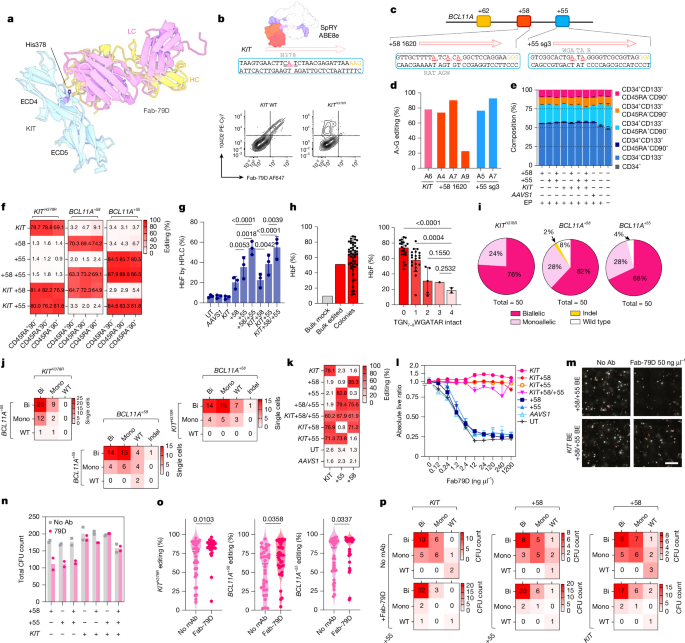
MainHSPC transplantation (HSCT) is a cornerstone therapy for a wide range of malignant and non-malignant conditions, leveraging the unique regenerative capacity of HSPCs to replenish the haematopoietic system14. Each year, more than 8,000 individuals undergo allogeneic HSCT in the USA, with even more receiving autologous transplants3,14,15. The therapeutic potential of HSCT has been further expanded by advancements in gene therapy, which entail the genetic modification of host HSPCs to ameliorate disease phenotypes16. However, HSCT carries substantial early and late treatment-related morbidity and mortality1,2,3,4. Even in patients who surviving more than five years after transplant, mortality remains fourfold to ninefold higher than in the general population3,17. The toxicities associated with conditioning regimens, which rely on high-dose chemo- and radiotherapy, are major contributors to these risks18,19. Myeloablative agents clear the haematopoietic stem cell (HSC) niche and, in allo-HSCT, contribute to host immunosuppression to overcome immunological barriers2,20. However, non-specific genotoxicity has both immediate and long-lasting effects18,19, including multi-organ damage (to lung, liver, kidney and nervous system) and haematopoietic aplasia, leading to infections, anaemia and bleeding. Survivors face lifelong risk of secondary malignancies, endocrine dysfunction and infertility. Furthermore, inflammation triggered by genotoxic conditioning may increase the risk of graft-versus-host disease21,22. These effects disproportionately affect frail individuals with multiple comorbidities and those with advanced malignancies, thereby constraining the broader applicability of HSC therapies.To overcome these challenges, targeted immunotherapies have been repurposed as safer alternatives to conventional regimens. Monoclonal antibodies (mAbs)5,6,7, antibody–drug conjugates (ADCs)23 and chimeric antigen receptor (CAR)-T cells24,25 that recognize the HSC marker KIT (also known as CD117) have been investigated to selectively deplete endogenous HSPCs26,27,28. Such approaches minimize organ damage, shorten aplasia and decrease pro-inflammatory mediators, sparing bone marrow and thymic niches, enabling faster immune recovery29. These strategies are particularly suitable for autologous gene therapies, where immune-suppression or complete haematopoietic replacement are not necessarily required. However, immune-based agents pose efficacy and safety challenges due to nonselective targeting of transplanted HSPCs and prolonged half-life, leading to on-target depletion30,31. Consequently, transplantation must be delayed until the drug is cleared or sufficiently inactivated. This delay can result in competition between transplanted and host HSPCs, resulting in lower chimerism compared with conventional conditioning30. To ensure time-limited HSPC depletion, ADCs with accelerated pharmacokinetics have been used, but these may increase damage to organs that metabolize the toxic payload32. Although in certain settings corrected cells have a selective advantage that may compensate for lower engraftment levels29,33, most genetic diseases require a minimum threshold for correction. For instance, β-haemoglobinopathies, prevalent blood disorders caused by mutations in the β-globin gene (HBB), require more than 20% correction for therapeutic benefit34,35.We recently showed that precise editing of target epitopes within donor HSPCs confers selective resistance to mAbs or CAR-T cells, without compromising protein function or regulation36. Here we leverage this technology to develop a clinically applicable platform for selective engraftment and progressive enrichment of therapeutically gene-modified HSPCs by coupling non-genotoxic immunotherapy-based myeloablation with epitope engineering.Multiplex editing of KIT and BCL11AWe previously36 identified mutations in KIT extracellular domain 4 (ECD4) that abrogate the binding of Fab-79D, a mAb that blocks SCF-induced KIT homodimerization8. Conversion of histidine 378 to arginine (Fig. 1a and Extended Data Fig. 1a) can be achieved in primary human mobilized peripheral blood-derived CD34+ HSPCs by adenine base editing (ABE), leading to permanent loss of mAb recognition (Fig. 1b) while preserving functionality, engraftment and differentiation capacity36. As ABE is well-suited for multiplexing, we combined epitope editing with disruption of the erythroid-specific BCL11A enhancer (+58 and +55 sites; Fig. 1c), a strategy to enforce HbF de-repression and improve sickle cell disease (SCD) and transfusion-dependent β-thalassaemia phenotypes12,13,37. A similar SpCas9 nuclease strategy (exagamglogene autotemcel (Casgevy)) has recently received approval from the US Food and Drug Administration38. After electroporation optimization (Extended Data Fig. 1b), mutiplex base editing of KITH378R, BCL11A+58 and BCL11A+55 resulted in efficient editing (78%, 72% and 74%, respectively; Fig. 1d–k and Extended Data Fig. 1c–e), with levels only slightly lower than for individual editing (Fig. 1k). KITH378R plus BCL11A+58/+55 triplex base editing did not affect the phenotypic composition of CD34+ HSPCs (Fig. 1e), with similar editing across progenitors sorted by fluorescence-activated cell sorting (FACS) and stem-enriched subsets (CD34+CD133+CD45RA−CD90+; Fig. 1f and Extended Data Fig. 1h). In vitro erythroid differentiation confirmed induction of HbF in BCL11A-edited cells (Fig. 1g and Extended Data Fig. 1f). Similar to nuclease-mediated disruption38, dual +58 and +55 targeting by ABE resulted in superior HbF induction compared with individual edits (Fig. 1g). Single-cell FACS confirmed that HbF expression was proportional to the number of disrupted core-half E-box/GATA motifs (TGN7–9WGATAR; Fig. 1h). Genomic analyses revealed a large fraction of biallelic edits (78%, 62% and 68% for KITH378R, BCL11A+58 and BCL11A+55, respectively; Fig. 1i), which translated to a high rate of KITH378R and BCL11A+58/+55 co-editing in single cells (Fig. 1j), providing the basis for co-selection.Fig. 1: Multiplex KIT and BCL11A base editing enables HbF induction and supports mAb-based selection.a, Structural model (adapted from Protein Data Bank (PDB): 4K9E) of KIT ECD4 bound to Fab-79D. His378 is indicated. HC, heavy chain; LC, light chain. b, Top, strategy to introduce the H378R mutation by ABE in exon 7 of KIT (structure adapted from PDB: 6VPC). Bottom, representative flow cytometry of bone marrow from NBSGW mice engrafted with unedited or KITH378R base-edited CD34+ HSPCs stained with Fab-79D and control clone 104D2. c, Base editing at the +58 and +55 DNase I hypersensitive sites within the BCL11A enhancer. Target edits within WGATAR motifs are highlighted in red. d, Representative base-editing efficiencies for KITH378R and BCL11A+58/+55 multiplex editing in HSPCs (n = 1). e, Phenotypic analysis of edited CD34+ HSPCs. Stacked bars show composition according to CD34, CD133, CD45RA and CD90 expression. Data are mean ± s.d. n = 3 donors. EP, electroporation. f, Heat maps of editing outcomes across sorted stem and progenitor subsets (CD45RA+CD90−, CD45RA−CD90− and CD45RA−90+) by editing combinations (left) and sequencing targets (top). n = 3 donors. g, HbF quantification by HPLC following single or multiplex edits. One-way ANOVA, Q < 0.05 indicated. Data are mean ± s.d. n = 3 donors. UT, untreated. h, HbF quantification after erythroid differentiation of single-cell clones according to the number of disrupted TGN7–9WGATAR motifs. One-way ANOVA, Q < 0.05 indicated. Data are mean ± s.d., n = 50 clones. i,j, Single-cell genotyping of edited CD34+ HSPCs, Distribution of monoallelic (Mono), biallelic (Bi) and indel outcomes for KITH378R, BCL11A+58 and BCL11A+55 (i) and allelic composition heat maps showing frequent biallelic co-editing (j). n = 50 cells. WT, wild type. k,l, In vitro selection of multiplex-edited HSPCs using Fab-79D antibody. Baseline editing efficiencies (k; n = 3 donors) and normalized live CD34+ counts after 7-day culture across mAb doses (l; normalized to dose = 0; data are mean ± s.d. n = 4 replicates). m,n, CFU assays plated with or without 50 ng µl−1 Fab-79D. Representative images (m) and CFU quantification (n; n = 2; editing conditions as indicated). Scale bar, 5 mm. BE, base editing. o,p, Single-colony genotyping. Unpaired t-test (P < 0.05 indicated) (o) and allelic composition heat maps of KITH378R, BCL11A+58 and BCL11A+55 with or without Fab-79D (p). Two-sided t-test.Source dataTo characterize their immune resistance, combinations of KIT, BCL11A+58 and BCL11A+55 base-edited HSPCs (Fig. 1k) were cultured in SCF-containing medium with different Fab-79D concentrations. After 7 days, KIT-unedited conditions showed significant growth inhibition, whereas KITH378R cells—alone or combined with BCL11A base-edited cells—expanded similarly to untreated cells (Fig. 1l). This protection matched that conferred by SpCas9-mediated KIT knockout, which reduced expansion but rendered cells unresponsive to mAb-mediated selection (Extended Data Fig. 1g). Similarly, colony-forming unit (CFU) assays of triplex-edited CD34+ HSPCs plated with or without Fab-79D confirmed growth inhibition of KIT-unedited colonies (Fig. 1m,n). Genomic analysis of individual colonies revealed significant co-occurrence of KIT and BCL11A editing before mAb-mediated selection (Supplementary Table 1). Consistently, exposure to Fab-79D co-enriched editing across all three loci (Fig. 1o). The allelic composition of Fab-79D-treated colonies showed selection of biallelic edits not only for KIT but also for BCL11A+58 and BCL11A+55 loci (Fig. 1p). These results confirm that base editing-mediated disruption of HbF-repressive elements can be combined with epitope editing in a single ex vivo manipulation, and that multiplex-edited HSPCs can be enriched by antagonistic anti-KIT mAbs.
Non-genotoxic transplantation and in vivo selection through epitope editing - Nature
Epitope editing of KIT enables antibody-based, non-genotoxic conditioning that selectively enriches therapeutic BCL11A-edited haematopoietic stem/progenitor cells, supports durable engraftment, preserves clonal diversity and enhances induction of fetal haemoglobin, a therapeutic approach for conditions such as sickle cell disease and β-thalassemia.
20,089 words~91 min read