You have full access to this article via your institution.
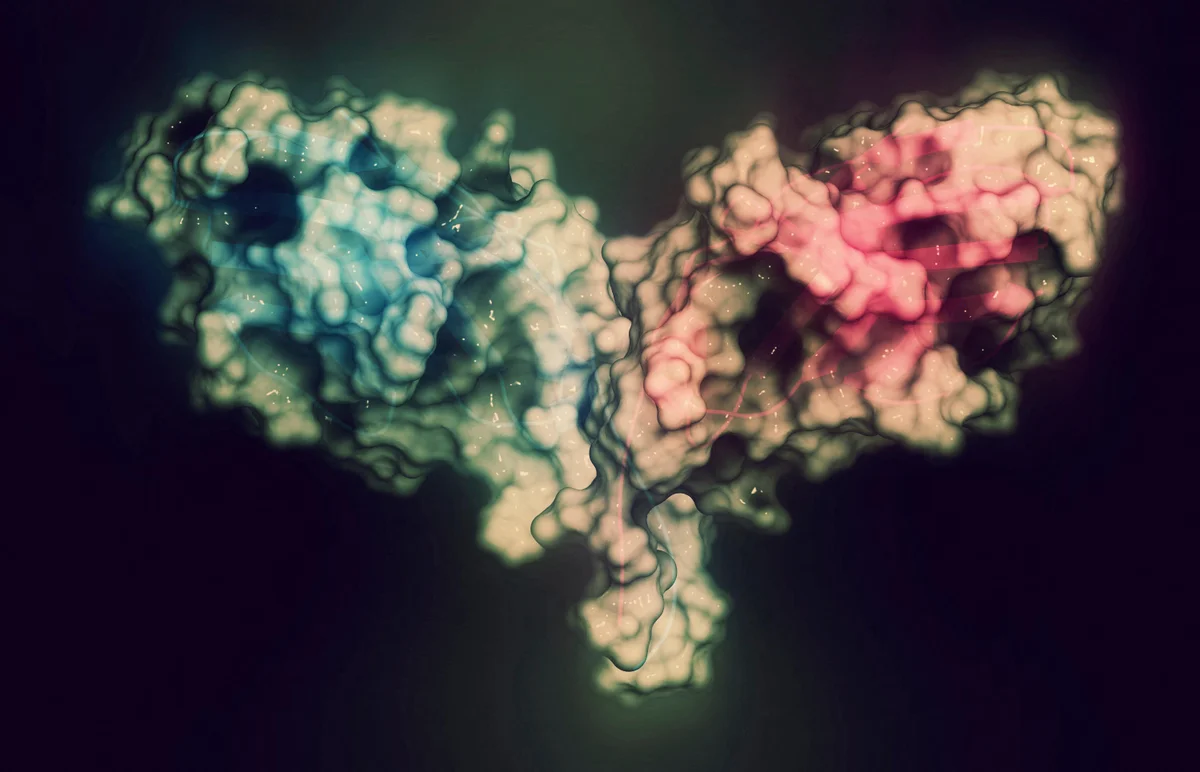
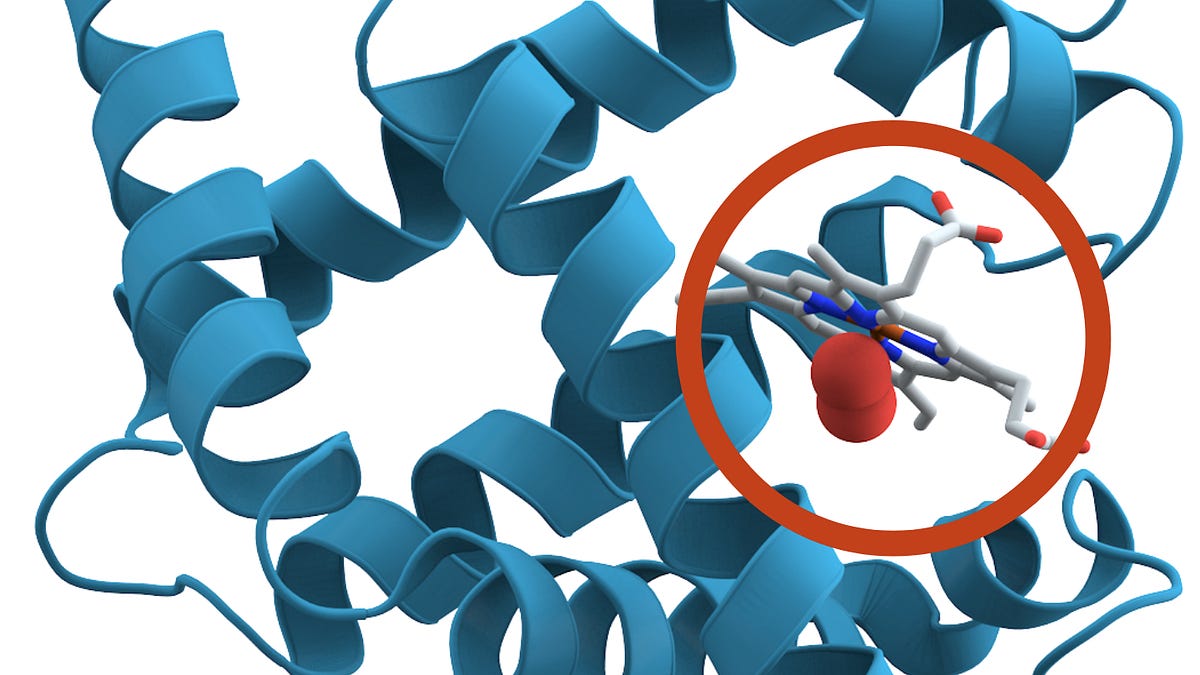
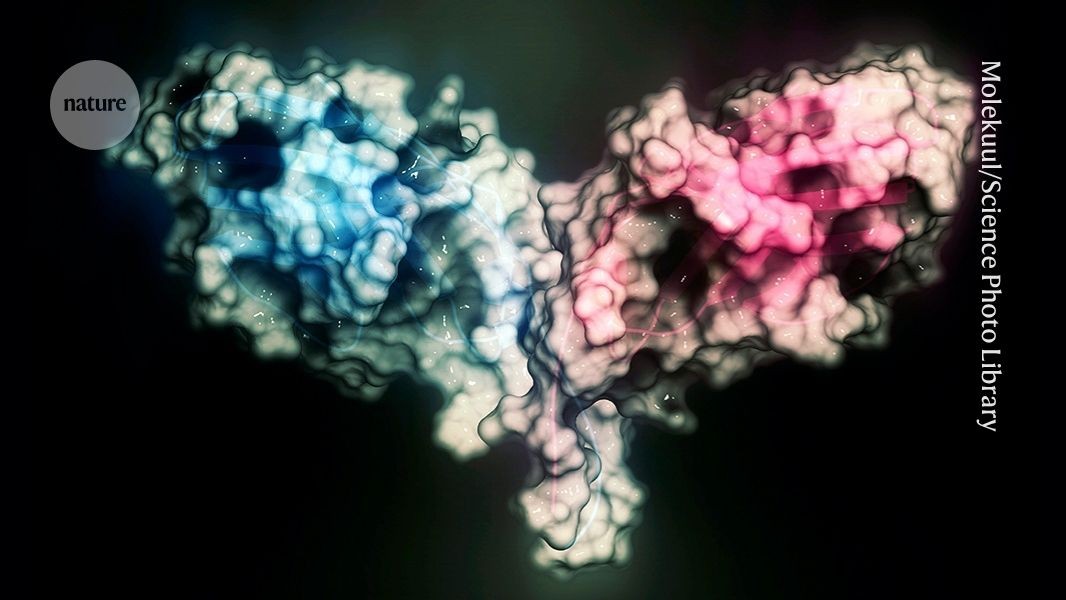
The AI tool designed binders against Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4). Credit: Molekuul/Science Photo LibraryThe known protein universe just got a lot bigger. A newly released artificial-intelligence tool has generated an atlas of more than one billion predicted protein structures and billions more protein sequences.The database, known as the ESM Atlas, was unveiled today by researchers at the Chan Zuckerberg Initiative’s Biohub (CZI-Biohub), a biomedical institute created in San Francisco, California, by Facebook founder Mark Zuckerberg and his wife, physician and educator Priscilla Chan.The atlas eclipses the AlphaFold Database of predicted protein structures by more than 800 million entries, and a previous ESM Atlas by some 300 million.The predictions were made using ESMFold2, an AI model that CZI-Biohub says surpasses the performance of AlphaFold3, the latest version of Google DeepMind’s system and other protein-structure prediction AIs. The atlas is described in a preprint released today1.“What this atlas does is it shows the totality of protein biology and especially the parts that are most unknown,” says CZI-Biohub science head Alex Rives, who led the effort. “We think it’s going to be a really powerful substrate for the discovery of new biology.”Other scientists are impressed with the results, especially that ESMFold2 is fully open source. But the CZI-Biohub model enters an increasingly crowded field, in which competing open-source and proprietary protein models are making gains at breakneck speed.Antibody predictionsESMFold2 is based on a ‘protein language’ model that Rives’s team unveiled in 2024, which was trained on billions of proteins from across the tree of life. It includes ‘metagenomic’ sequences from soil, ocean and other environments, which are absent from the AlphaFold database of predicted protein structures.Rives’ team say ESMFold2 outperforms existing methods, including AlphaFold3, at determining the correct structure of complexes of interacting proteins – including antibody molecules binding to their antigen molecular targets.AlphaFold is running out of data — so drug firms are building their own versionIn the preprint, the researchers describe how they used ESMFold2 to design new antibodies and other proteins that can strongly attach to proteins implicated in cancers and immunological conditions. When created and tested in the lab, a high proportion of the designs worked as predicted.Rives’s team used the tool to create an atlas containing 1.1 billion predicted protein structures as well as information on the sequences of 6.8 billion proteins. Most of these come metagenomic sequences that had been only poorly characterized. Rives hopes that the atlas — which will be freely accessible — will help scientists to make connections between the known and unknown parts of the protein universe. Using the atlas, the researchers found structural similarities between CRISPR microbial defence proteins and a gene-editing protein identified in a soil fungus in 2023 and found in other eukaryotic species.Supplementary databaseThe newly released atlas should be “an extraordinary resource for biology,” says Gemma Atkinson, a computational biologist at Lund University in Sweden. “It's exciting to see how large scale protein language models can capture fundamental rules of protein biology.”Computational biologist, at University College London, says the predictions, which will first need evaluating, could help uncover new protein folds and functions, with implications for protein design and basic understanding of biology. Martin Steinegger, a computational biologist at Seoul National University, says his biggest question is how well ESMFold2 can predict the structure of proteins that are very difference from those already known. His team found that the first edition of ESMFold wasn’t especially good at predicting unusual protein structures, especially those found in metagenome data2.Computational biologist Sergey Ovchinnikov at the Massachusetts Institute of Technology in Cambridge sees the ESM Atlas as a supplement to the widely used AlphaFold database of more than 200 million protein structures, rather than as a replacement.AlphaFold touted as next big thing for drug discovery — but is it?ESMFold2’s predictions of interacting proteins are impressive, Ovchinnikov adds, but not all that surprising. Earlier this year, the Google DeepMind biopharma spin-off Isomorphic Labs unveiled a proprietary model that made substantial gains at predicting such structures. Open-source models that the CZI-Biohub team didn’t compare ESMFold2 against directly have also achieved impressive results at predicting protein interactions, Ovchinnikov says.The fully open-source nature of ESMFold2, with no restrictions on commercial use, means that it could find wide use, says Ovchinnikov. “I expect many people will be excited to try ESMFold2.”